随着癌症的空间转录组分析的爆火,越来越多的肿瘤学TOP期刊收录了相当一部分的生信分析文章,很多都则用到了MIA(Multimodal intersection analysis)分析,这种分析方法小果还没有给大家介绍过,这让小果眼馋的同时也对这种新的富集分析方法怀有了好奇心,所以小果马上搜集了一篇Cancer reserch上的文章,带着小伙伴们一起学习并完成文章中的MIA分析的复现!本次的数据可能比较多,欢迎小伙伴们租赁我们的服务器来运行代码哦~

首先,小果隆重给大家介绍一下今天的核心方法——MIA分析,Multimodal intersection analysis,也就是多模式相交分析,是一种空间转录组和单细胞转录组整合分析的方法,用于评估特定组织区域中特定细胞类型的富集程度。其原理是计算空间转录组的区域差异基因与单细胞转录组鉴定的细胞类型的marker基因之间的重叠关系,使用超几何分布推断P值。MIA分析可以应用于不同的生物学问题,例如胰腺癌以及卵巢癌的初步分析种,可以利用MIA分析进行富集,对细胞的不同类型进行“拆分”评估,这使得MIA分析在疾病诊断和临床实践中也逐渐得到了越来越多的关注和应用。
那么应该怎样实现呢?小果这就以文献提供的数据和代码为例为小伙伴们进行说明。
在复现之前,需要搭建合适的工作环境,以下是文献所使用的详细配置:
R 4.1.0
Seurat 3.2.1
dplyr 1.0.9
limma latest
scales 1.2.0
imager 0.42.13
RColorBrewer 1.1.3
ggplot2 3.3.6
gplots 3.1.3
tidyverse 1.3.1
搭建好自己的平台之后,我们就可以进行数据分析了!如果大家不会搭建自己的平台,欢迎租赁我们的服务器哦~
第一步,将数据矩阵加载到Seurat中
本次复现所用的数据小果已经帮大家整理好了呦:
链接:https://pan.baidu.com/s/10mos4vmEvXunU1o3SQOz9g
提取码:ikl5
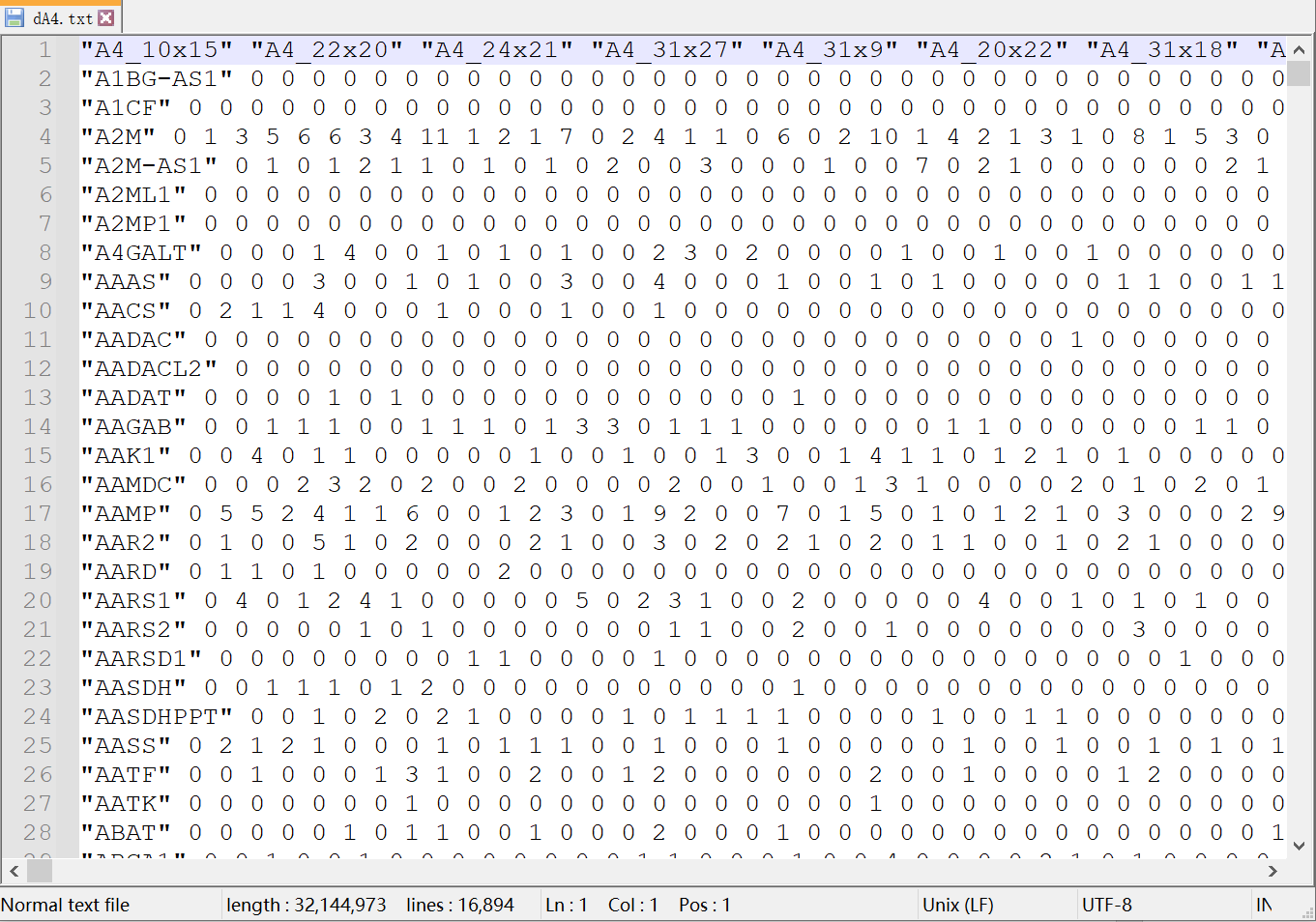
输入文件如图所示,是一个存储有空间转录组数据的文件,它包含了细胞的空间坐标和基因表达矩阵。
##1.导入包
library(Seurat)
library(dplyr)
library(limma)
#设置工作目录
data_dir = ‘/data/ST_data_matrices’
save_dir = ‘/results’
files = list.files(data_dir)
#读取数据
files <- c(“file1.txt”, “file2.txt”, “file3.txt”, “file4.txt”)
for (i in 1:4) {
file_path <- paste0(data_dir, “/”, files[i])
assign(paste0(“dA”, i), read.delim2(file_path, sep = ” “))
}
#将数据导入Seurat,进行PCA,UMAP分析
# 用一个向量存储数据框名
data_frames <- c(“dA4”, “dA5”, “dA10”, “dA12”)
# 用一个向量存储颜色名
colors <- list(c(‘orange’,’paleturquoise4′, ‘red’, ‘orchid’, ‘seagreen2’, ‘darkgreen’, ‘slateblue1′,’yellow’,’pink’),
c(‘orange’,’paleturquoise4′, ‘orchid’, ‘seagreen2′,’slateblue1′,’red’,’yellow’),
c(‘orange’,’paleturquoise4′,’red’,’darkgreen’,’orchid’,’blue’,’cyan’),
c(‘red’,’darkgreen’, ‘orange’,’paleturquoise4′, ‘orchid’,’blue’))
# 用一个循环对每个数据框进行分析
for (i in 1:4) {
sample <- CreateSeuratObject(counts = get(data_frames[i]), project = “ST_HGSOC”, min.cells = 2, min.features = 200)
#数据进行标准化和变换
sample <- SCTransform(sample, verbose = FALSE, return.only.var.genes = FALSE)
#主成分分析
sample <- RunPCA(sample, assay = “SCT”, verbose = FALSE) # return top 50 PCs
#聚类分析
sample <- FindNeighbors(sample, reduction = “pca”, dims = 1:10) %>%
FindClusters(verbose = FALSE) # default k.param = 30 for the k-nearest neighbor algorithm
#进行降维和可视化
sample <- RunUMAP(sample, reduction = “pca”, dims = 1:10) %>%
DimPlot(reduction = “umap”, label = F, cols = colors[[i]])
}
## 2.查找DEG(差异表达基因)并绘制热图,利用UMAP绘制簇
#计算每个样本中的平均基因
# data=sample12@assays$RNA
# temp = 0
# for (i in seq(1,dim(data)[2]))
# {temp = temp+sum(data[,i]==0)}
# avg1 = dim(data)[1]-temp/dim(data)[2]
## 3.保存Seurate文件
fnm = paste0(save_dir,’/seurat_object.RData’)
save(sample4,sample5,sample10,sample12, file = fnm)
## 4.保存每个簇的差异表达基因
folder = paste0(save_dir,’/ST differentially expresssed genes up and down’)
if (!file.exists(folder)){
dir.create(folder)
}
samples = c(sample4,sample5,sample10,sample12)
sample_names = c(‘A4′,’A5′,’A10′,’A12′)
for (j in seq(1,length(samples))){
tmp1 = samples[[j]]
fnm_pt1 = sample_names[j]
tmp = tmp1@meta.data$seurat_clusters
n_clu = nlevels(tmp)
for (i in seq(0,n_clu-1,by = 1)){
fnm = paste0(folder,’/’,fnm_pt1,’_c’,i,’.xls’)
tmp_marker <- FindMarkers(tmp1, ident.1 = i, min.pct = 0.25, only.pos = FALSE,logfc.threshold = 0)
tmp_marker$gene <- rownames(tmp_marker)
tmp_mk <- tmp_marker %>% select(gene, everything())
tmp_mk <- tmp_mk[order(-tmp_mk$avg_logFC),]
tmp_mk <- tmp_mk[tmp_mk$p_val<0.05,]
write.table(tmp_mk,fnm,quote=F,sep=”\t”,row.names = F)
}
}
## 5.保存每个簇的数量
find_cluster_numbers <- function(SOE, SID) {
SOE@meta.data %>%
count(seurat_clusters) %>%
mutate(name = paste0(SID, “_c”, seurat_clusters)) %>%
select(name, n) %>%
column_to_rownames(“name”)
}
ss <- bind_rows(
find_cluster_numbers(sample4, “A4”),
find_cluster_numbers(sample5, “A5”),
find_cluster_numbers(sample10, “A10”),
find_cluster_numbers(sample12, “A12”)
)
colnames(ss) <- “spots”
# 创建一个文件夹来保存结果
folder <- file.path(save_dir, “ligand_receptor”)
dir.create(folder, showWarnings = FALSE)
file_name <- file.path(folder, “ligand_receptor_node_attribute.csv”)
write.csv(ss, file_name)
下图是我们聚类之后的细胞簇分布数据,文件存储了每个细胞簇的节点属性,其中第一列是细胞簇的名称,而第二列是细胞簇中的细胞数。我们以第一个样本为例,有8个细胞簇,分别命名为A4_c0到A4_c7,其中的细胞数从27到240不等,这为我们后续的MIA分析打下了基础。
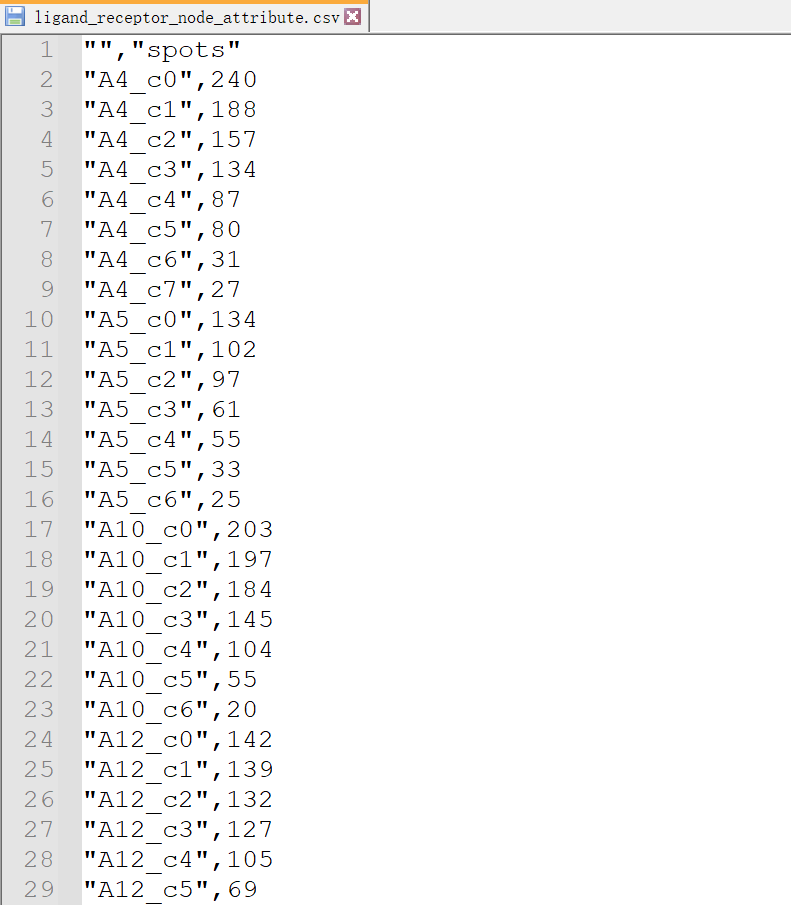
ligand_receptor_node_attribute.csv
第二步,将Seurat对象中的聚类结果映射到细胞的空间位置中
Seurat包是进行生信分析非常重要的工具,小果在之前的文章中已经和大家介绍过Seurat的详细使用方法了,小伙伴们如果有需要,可以关注公众号进行搜索哦。
#导入包
library(ggplotify)
library(ggplot2)
library(magick)
library(scales)
library(imager)
save_dir = ‘/results’
image_dir = ‘/data/images’
fnm = paste0(save_dir,’/seurat_object.RData’)
load(fnm)
#绘制无背景的聚类结果
plot_clu_nbg <- function(SID,SOE,d,topleft,adjust_x,adjust_y,delta_x,delta_y,im){
tmp = SOE@meta.data$seurat_cluster
Nclu = length(levels(tmp))
nspots = length(tmp)
colorcode = hue_pal()(Nclu)
colorcode = c(‘orange’,’green’,’blue’,’red’,’cyan’,’magenta’,’yellow’,’brown’)
mycols <- c(rep(NA,length(tmp)))
for (i in seq(1,nspots)){
mycols[i] = colorcode[tmp[i]]
}
filenm = paste0(folder,’/A’,SID,’noBG.png’)
png(filenm, width = 512, height = 512)
par(mar = c(0,0,0,0)) # set zero margins on all 4 sides
plot(x = NULL, y = NULL, xlim = c(0,dim(im)[1]), ylim = c(0,dim(im)[2]), pch = ”,
xaxt = ‘n’, yaxt = ‘n’, xlab = ”, ylab = ”, xaxs = ‘i’, yaxs = ‘i’,
bty = ‘n’) # plot empty figure
# rasterImage(im, xleft = 0, ybottom = 0, xright = dim(im)[1], ytop = dim(im)[2])
#解除本行注释即可绘制有背景的聚类图
points(topleft[1]+(d[,1]-1)*delta_x+apply(d,1,function(x){return(adjust_x(x[2]))}),
topleft[2]-(d[,2]-1)*delta_y+apply(d,1,function(x){return(adjust_y(x[1]))}),
col=mycols,pch=20,cex=2.8)
dev.off()
}
#绘制每个样本的簇图
plot_clu <- function(SID, SOE, d, topleft, adjust_x, adjust_y, delta_x, delta_y, im) {
tmp <- SOE@meta.data$seurat_cluster
colorcode <- c(‘orange’,’green’,’blue’,’red’,’cyan’,’magenta’,’yellow’,’brown’)
df <- data.frame(
x = topleft[1] + (d[, 1] – 1) * delta_x + apply(d, 1, function(x) adjust_x(x[2])),
y = topleft[2] – (d[, 2] – 1) * delta_y + apply(d, 1, function(x) adjust_y(x[1])),
col = colorcode[tmp]
)
filenm <- file.path(folder, paste0(“A”, SID, “.png”))
ggplot(df, aes(x, y, color = col)) +
annotation_raster(im, xmin = 0, xmax = dim(im)[1], ymin = 0, ymax = dim(im)[2]) +
geom_point(size = 1.7) +
theme_void() +
guides(color = FALSE) +
ggsave(filenm, width = 512, height = 512, units = “px”)
}
# 读取图像文件
fnm <- file.path(image_dir, “Sample_4_ds.jpg”)
im <- image_read(fnm)
# 将图像转换为ggplot对象
p <- as.ggplot(im)
# 使用ggplot2的功能来修改图像
p + ggtitle(“Sample 4”) + theme(plot.title = element_text(hjust = 0.5))
#将所有聚类得到的簇绘制在背景上
plot_clu(SID,SOE,d,topleft,adjust_x,adjust_y,delta_x,delta_y,im)
plot_clu_nbg(SID,SOE,d,topleft,adjust_x,adjust_y,delta_x,delta_y,im)
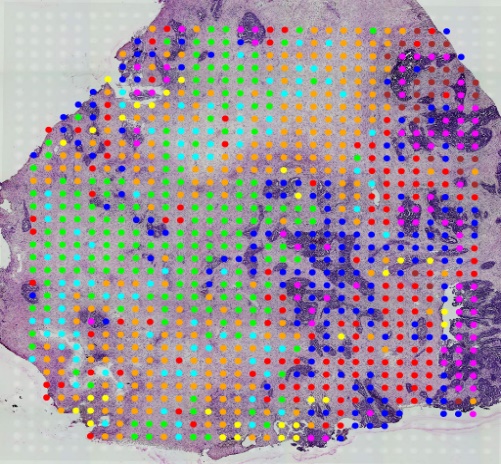
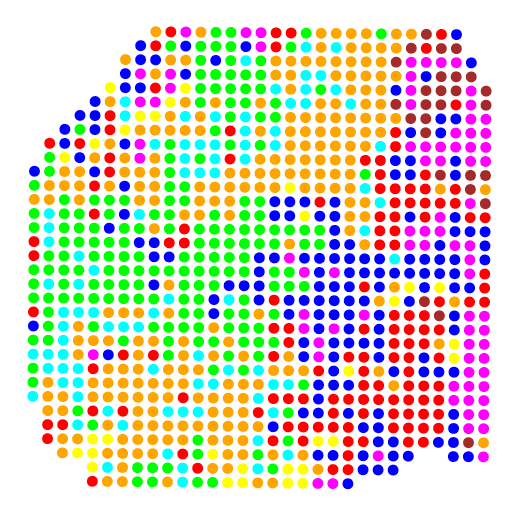
A4.png
A4noBG.png
如图,我们将来自卵巢癌患者样本的空间转录组数据与公开可用的单细胞RNA测序数据进行了整合。并将每个空间转录组组织冷冻切片都安装在一个空间编码的微阵列上。微阵列上的斑点根据多个区域的基因表达进行了测序和聚类。单细胞RNA测序数据也进行了聚类,生成了多种细胞类型及其特异性基因集。
通过这一步映射,就可以清晰的利用不同的颜色区分特异的基因集了,而且绘制出的图片也是非常有对照特性的。
第三步,设定scRNA的特征基因
#导入包
library(ComplexHeatmap)
library(stringr)
library(RColorBrewer)
data_dir = ‘/data/scRNAseq_data’
save_dir = ‘/results’
folder = paste0(save_dir,’/scRNAseq’)
if (!file.exists(folder)){
dir.create(folder)
}
#从 scRNAseq Shih et al PLoS ONE 13(11): e0206785下载数据
library(purrr)
# 读取,转置数据
read_transpose <- function(file) {
data <- read.csv(gzfile(file), check.names = FALSE)
data <- t(data)
colnames(data) <- data[1, ]
data <- data[-1, ]
class(data) <- “numeric”
data
}
HG <- c(
“GSM3348305_589_Omentum_S1.counts.umiCounts.aboveBackground.table.csv.gz”,
“GSM3348309_TB10040580_S1.counts.umiCounts.table.csv.gz”,
“GSM3348310_TB10040580met_S1_1_.counts.umiCounts.table.csv.gz”,
“GSM3348311_TB10040589_4_S1.counts.umiCounts.table.csv.gz”,
“GSM3348312_TB10040589met_S1.counts.umiCounts.table.csv.gz”,
“GSM3348313_TB10040600_CD31_S1.counts.umiCounts.table.csv.gz”,
“GSM3348314_TB10040600_NP-1_S1.counts.umiCounts.table.csv.gz”,
“GSM3348315_TB10040600_NP-2_S1.counts.umiCounts.table.csv.gz”,
“GSM3348316_TB10040600_Tumor_S1.counts.umiCounts.table.csv.gz”,
“GSM3348319_V00331081_Primary_S1.counts.umiCounts.aboveBackground.table.csv.gz”,
“GSM3348320_V00331151_Metastatic_S1.counts.umiCounts.aboveBackground.table.csv.gz”
)
#设置工作目录
setwd(file.path(data_dir, “GSE118828_RAW”))
data <- map_dfr(HG, read_transpose, .id = “file”)
#将数据加载到 Seurat、Clustering、Cluster Annotation 中
#tmp.index = grep(“_”,rownames(data))
#rownames(data)[tmp.index] = lapply(rownames(data)[tmp.index],str_replace,pattern=”_”,replacement=”-“)
D13_data <- CreateSeuratObject(counts = data, project = “HGSOC”, min.cells = 3, min.features = 200)
D13_data[[“percent.mt”]] <- PercentageFeatureSet(D13_data, pattern = “^mt-“)
VlnPlot(D13_data, features = c(“nFeature_RNA”, “nCount_RNA”, “percent.mt”), ncol = 3)
D13_data <- subset(D13_data, subset = nFeature_RNA > 500 & nFeature_RNA < 10000 & percent.mt < 25)
D13_data <- NormalizeData(D13_data, normalization.method = “LogNormalize”, scale.factor = 10000)
D13_data <- FindVariableFeatures(D13_data, selection.method = “vst”, nfeatures = 3000)
#为所有簇绘制热图并进行质量控制
data0 <- D13_data@assays$RNA@counts
D13_data <- CreateSeuratObject(counts = data0, min.cells = 3, min.features = 200)
D13_data <- NormalizeData(D13_data)
D13_data <- FindVariableFeatures(D13_data, selection.method = “vst”, nfeatures = 2000)
VizDimLoadings(D13_data, dims = 1:4, reduction = “pca”)
D13_data <- RunPCA(D13_data)
ElbowPlot(D13_data)
# 寻找相邻细胞
D13_data <- FindNeighbors(D13_data, dims = 1:10)
# 寻找细胞簇
D13_data <- FindClusters(D13_data, resolution = 0.5)
# 运行UMAP
D13_data <- RunUMAP(D13_data, dims = 1:10)
# 可视化UMAP结果
DimPlot(D13_data, reduction = “umap”, pt.size = 0.5)
ctypes <- c(“Ep_1”, “FB_1”, “T”, “Ep_2”, “Myeloid”, “Ep_3”, “FB_2”, “Ep_4”, “Endothelial”, “B”, “FB_3”)
D13_data$ctypes <- factor(ctypes[D13_data$seurat_clusters])
Idents(D13_data) <- “ctypes”
DimPlot(D13_data, reduction = “umap”, pt.size = 0.5)
D13_data.markers <- FindAllMarkers(D13_data, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
D13_data.markers <- D13_data.markers[D13_data.markers$p_val < 10^(-5), ]
D13_data.markers$cell_type <- ctypes[D13_data.markers$cluster]
fnm <- file.path(folder, “scRNAseq_differentially_expressed_genes_shih_2018_annotated.xls”)
write.table(D13_data.markers, fnm, quote = FALSE, sep = “\t”, row.names = FALSE)
#为所有簇绘制热图
Tc.averages <- AverageExpression(Tc, return.seurat=TRUE)
temp = Tc.averages@assays$RNA@scale.data
fnm = paste0(folder,’/scRNAseq_differentially_expressed_genes_shih_2018.xls’)
DE = read.table(fnm,header = TRUE, stringsAsFactors = F, check.names=F)
top15 <- DE %>% group_by(cluster) %>% top_n(n = 15, wt = avg_logFC)
# DE = DE[DE$avg_logFC>1.8,]
genelist = top15$gene
# genelist = DE$gene
temp = temp[genelist,]
my_palette <- c(“black”, “white”, “brown”)
data <- data.matrix(temp)
ht <- Heatmap(data,
name = “scaled value”,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = FALSE,
show_column_names = TRUE,
column_names_rot = 60,
col = my_palette,
heatmap_legend_param = list(title_position = “topleft”, legend_width = unit(4, “cm”))
)
# 保存热图为pdf文件
filename <- file.path(folder, “scRNAseq differentially expresssed genes.pdf”)
pdf(filename, 8, 8)
draw(ht)
dev.off()
如图所示,是小果绘制的细胞类型特异性基因集,用于展示多个变量之间的关系。其中的红色代表较高的比例数值,白色代表中等数值,灰色代表较低的数值。下方的标签(T,B, Myeloid,Ep_1等等)表示的是不同的细胞类型或亚群,只是进行MIA分析所必需的数据。而右侧的标签(如CD81, MSX1等)表示的是选定的scRNA。红色区域表示细胞亚群和scRNA之间存在较强的相关性或相互作用。
例如,图中的T细胞和B细胞分别在CD3D和CD83基因上有较高的表达,而EP 1-1细胞和EP 1-2细胞在ATHL1基因上有较高的表达,进而反映了不同细胞类型或亚群的特征和功能,这样我们就可以非常明显的看到癌症病人的特异的scRNA了。
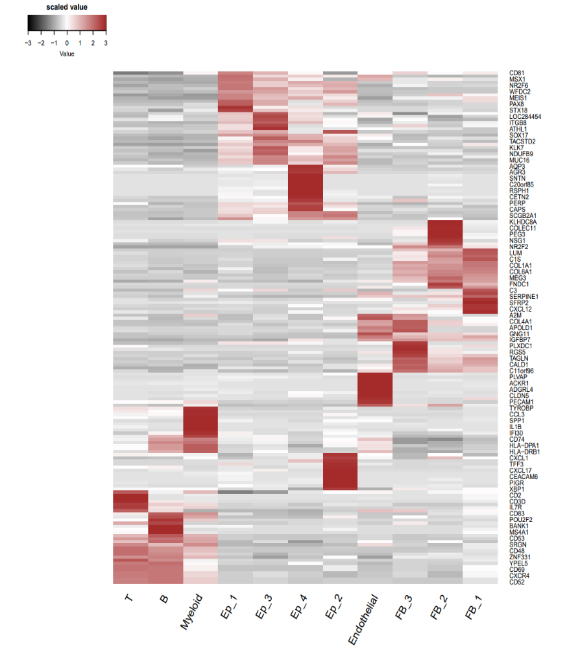
ScRNA特征基因的DEG
第四步,MIA分析
接下来是小果期待已久的MIA分析!
#导入包
library(tidyverse)
save_dir = ‘/results’
fnm = paste0(save_dir,’/seurat_object.RData’)
load(fnm)
folder = paste0(save_dir,’/MIA’)
if (!file.exists(folder)){
dir.create(folder)
}
##1.从 PLoS One. 2018; 13(11): e0206785 中加载 scRNAseq 的 DEG(差异表达基因)
load_scRNA_DEG <- function(){
fnm = paste0(save_dir,’/scRNAseq/scRNAseq_differentially_expressed_genes_shih_2018.xls’)
tmp1 = read.table(fnm,header = TRUE, stringsAsFactors = F, check.names=F)
scRNA = tmp1[tmp1$p_val<10^(-5),]
clusters = unique(scRNA$cluster)
n_sc = length(clusters)
scgenelist = list()
for (i in seq(1,n_sc)){
scgenelist[i] = list(scRNA[scRNA$cluster==clusters[i],”gene”])
}
return(scgenelist)
}
##2.加载 ST 数据的 DEG(差异表达基因)
load_ST_DEG <- function(SOE){
tmp = SOE@meta.data$seurat_clusters
n_st = nlevels(tmp)
stgenelist = list()
for (i in seq(0,n_st-1,by = 1)){
tmp_marker <- FindMarkers(SOE, ident.1 = i, min.pct = 0.25)
tmp_marker$gene <- rownames(tmp_marker)
tmp_mk <- tmp_marker %>% select(gene, everything())
tmp_mk <- tmp_mk[order(-tmp_mk$avg_logFC),]
tmp_mk <- tmp_mk[tmp_mk$p_val<0.01,]
tmp_mk <- tmp_mk[tmp_mk$avg_logFC>0,]
stgenelist[i+1] = list(tmp_mk$gene)
}
return(stgenelist)
}
##3.计算 MIA(多模态交叉分析)
# 定义一个函数来计算MIA值
compute_MIA <- function(scgenelist, stgenelist, SOE, SID) {
N_tot <- SOE@assays$RNA@counts@Dim[1]
MIA <- matrix(0, nrow = length(scgenelist), ncol = length(stgenelist))
MIA <- apply(MIA, c(1, 2), function(i, j) {
# 获取scRNA和ST的基因数
N_scRNA <- length(scgenelist[[i]])
N_ST <- length(stgenelist[[j]])
# 获取交集的基因数
N_intersect <- length(intersect(scgenelist[[i]], stgenelist[[j]]))
# 计算超几何分布的概率
P <- phyper(N_intersect, N_scRNA, N_tot – N_scRNA, N_ST, lower.tail = FALSE, log.p = FALSE)
# 计算MIA值
MIA_enrich <- -log10(P)
MIA_deplete <- -log10(1 – P)
if (MIA_enrich >= MIA_deplete) {
MIA_enrich
} else {
-MIA_deplete
}
})
MIA <- data.frame(MIA)
rownames(MIA) <- c(“Ep_1”, “FB_1”, “T”, “Ep_2”, “Myeloid”, “Ep_3”, “FB_2”, “Ep_4”, “Endothelial”, “B”, “FB_3”)
colnames(MIA) <- levels(SOE@meta.data$seurat_clusters)
# 保存结果
fnm <- paste0(folder, “/”, SID, “.xls”)
MIA <- format(MIA, digits = 3)
write.table(MIA, fnm, quote = FALSE, sep = “\t”, col.names = NA)
}
scgenelist <- load_scRNA_DEG()
SOE <- sample4
SID <- “A4”
stgenelist <- load_ST_DEG(SOE)
compute_MIA(scgenelist, stgenelist, SOE, SID)
##4.绘制MIA的热图
load_MIA <- function(SID){
fnm = paste0(folder,’/’,SID,’.xls’)
temp1 = read.table(fnm,header = TRUE, stringsAsFactors = F, check.names=F)
cnames = colnames(temp1)
cnames1 = paste0(SID,’_c’,cnames)
colnames(temp1) = cnames1
return(temp1)
}
#合并不同的簇
temp = load_MIA(‘A4’)
for (id in c(‘A5′,’A10′,’A12’)){
temp1 = load_MIA(id)
temp = merge(temp,temp1,by = 0)
rownames(temp) = temp[,1]
temp = temp[,-1]
}
#绘制热图
my_palette <- c(“red”, “black”, “cyan”)
data <- data.matrix(temp)
data[data > 40] <- 40
filename <- file.path(folder, “MIA_heatmap.pdf”)
#使用pheatmap绘制并保存热图
pheatmap(data,
filename = filename,
cluster_rows = FALSE,
cluster_cols = FALSE,
color = my_palette,
legend = TRUE,
legend_breaks = seq(0, 40, by = 5),
legend_labels = c(“0”, “5”, “10”, “15”, “20”, “25”, “30”, “35”, “40”),
cellwidth = 10,
cellheight = 6,
fontsize = 10,
fontsize_row = 10,
fontsize_col = 10,
angle_col = 90,
main = “P value (-log10)”,
xlab = “ST clusters”
)
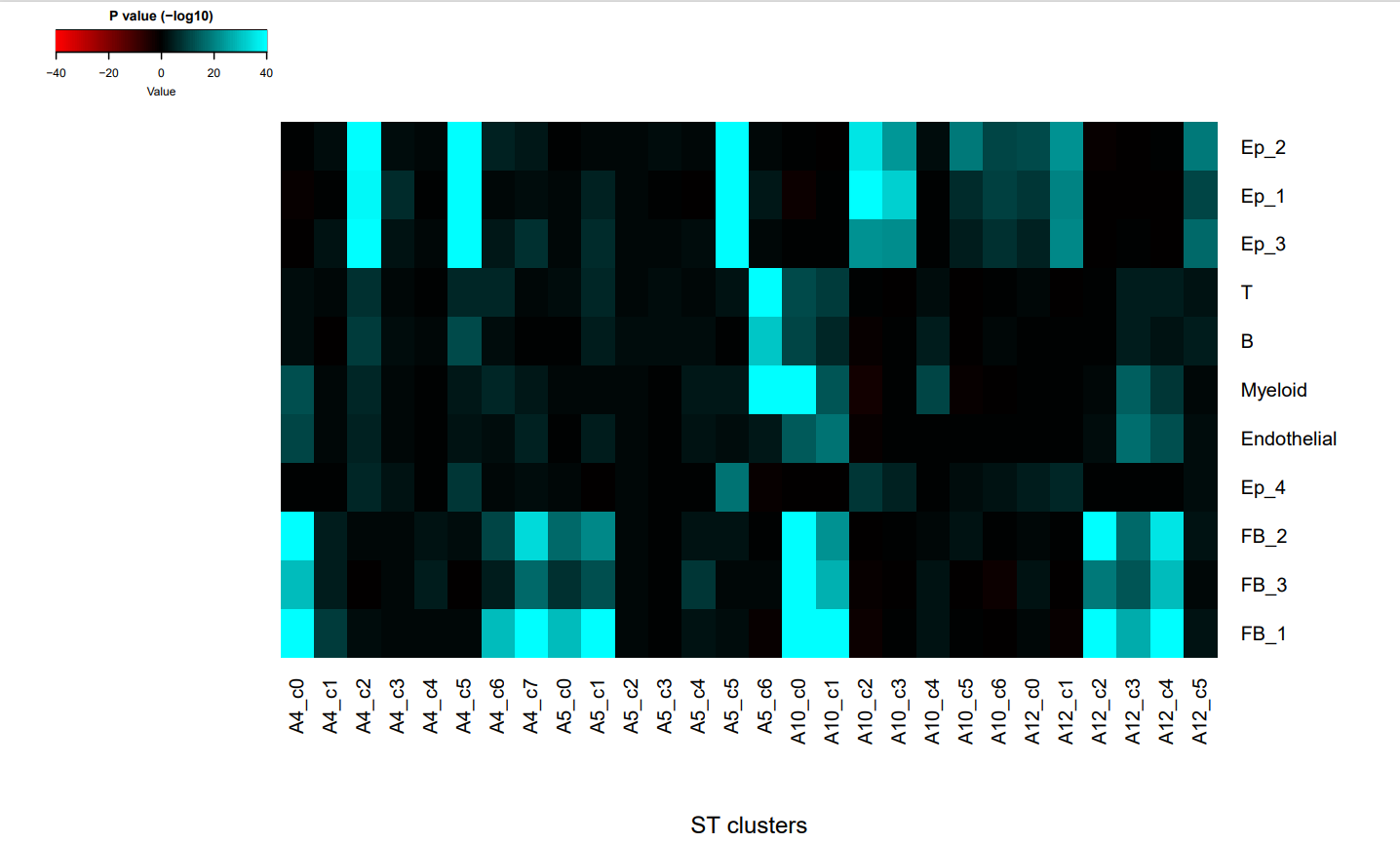
MIA分析热图
如图是小果绘制的MIA图,该图显示了所有单细胞RNA测序识别的细胞类型和空间转录组学定义的区域,并且表示了不同ST集群和细胞类型之间的P值。P值是用于衡量观察到的数据与假设之间的一致性的一个检测标准,P值越小,表示观察到的数据与假设之间的差异越大。
在这幅图中,用颜色条来表示P值的-log10转换,其中蓝色表示较高的P值,黑色表示较低的P值。图中的蓝色的区域表示ST集群和细胞类型之间有显著的差异,而黑色的区域表示ST集群和细胞类型之间没有显著的差异。例如Ep.2和Ep.3这两种细胞类型有很高的P值,说明它们与对照的正常细胞之间有很大的差异,而两个样本之间的P值又非常相似,说明这两个样本之间具有联系,很可能是我们通过MIA分析识别出来的肿瘤,也就是肿瘤簇,而FB_1和FB_2这两种细胞类型有很低的P值,说明它们和正常的细胞类型之间没有很大的差异,是正常细胞,也就是基质簇。这样我们就可以将肿瘤细胞的联系和正常的基质细胞的联系区分开来了。
小果今天的分享就到这里啦,在本次的教程中,我们学习了一种新的富集分析方法—MIA分析在癌症领域中的应用,通过对细胞进行富集分析进而“拆解”细胞类型,对肿瘤的空间异质性进行区分,这也是癌症分析的基础。小伙伴们如果没有做出来图也不要灰心,可以试一试我们的云生信小工具哟,只要输入合适的数据就可以直接绘制想要的图呢,链接:http://www.biocloudservice.com/home.html。