在生信分析的过程中,Seurat一直是我们的“传家宝”,是一个用于单细胞转录组数据分析的R语言包,可以帮助我们探索细胞的异质性和差异性。同时,别看它只是一个包,却提供了多种的符合功能,它提供了一套完整的流程,从数据导入、质控、归一化、特征选择、缩放、降维、聚类、可视化到差异表达分析等,都可以通过Seurat实现。本次运行的Seurat可能需要较多的服务器内存空间,如果小伙伴们自己的服务器不是很合适,欢迎小伙伴们租赁我们的服务器来运行代码哦~
而在热门的肿瘤分析领域,Seurat同样有着“统治性”的地位,小果今天就来教教大家,如何用Seurat实现肿瘤类文章的入门级分析—基因差异分析,从而将肿瘤细胞的差异基因进行区分,找到其中可能的“致命基因”。
首先,小果帮大家理清对肿瘤进行初步分析的思路,在获取肿瘤的相关数据后,我们需要对数据进行整合分析,评估特定组织区域中特定细胞类型的富集程度,然后再进行差异分析。
我们在反映基因的差异表达程度时,通常使用的是平均对数折叠变化(avg_logFC)。avg_logFC指的是两组样本(例如两种细胞类型或两种处理条件)之间基因表达量的比值的对数。对数折叠变化可以反映基因的差异表达程度,正值表示第一组样本中基因表达更高,负值表示第二组样本中基因表达更高。平均对数折叠变化是对每个基因在不同样本中的表达量取平均后再计算的对数折叠变化,常用于转录组分析或单细胞分析中,作为筛选差异表达基因的一个标准。
在了解如何分离差异表达基因之后,我们终于可以开始期待已久的差异分析了!
第一步:分离肿瘤基质簇
根据小果为大家提供的代码,首先导入所需的R包,这一步中主要用到的是Seurat和 scales,这两个包在往期的推送中有详细的介绍,小伙伴们可以关注小果并搜索哦。
本期所用的数据小果已经准备好放在下面的链接中了哟~
链接:https://pan.baidu.com/s/12y5rnPGjJT-oVYyCQzzELw
提取码:gbpc
输入数据如图所示,小果已经将所需的功能函数以及变量,单细胞转录组数据整理为R数据,大家直接导入即可使用 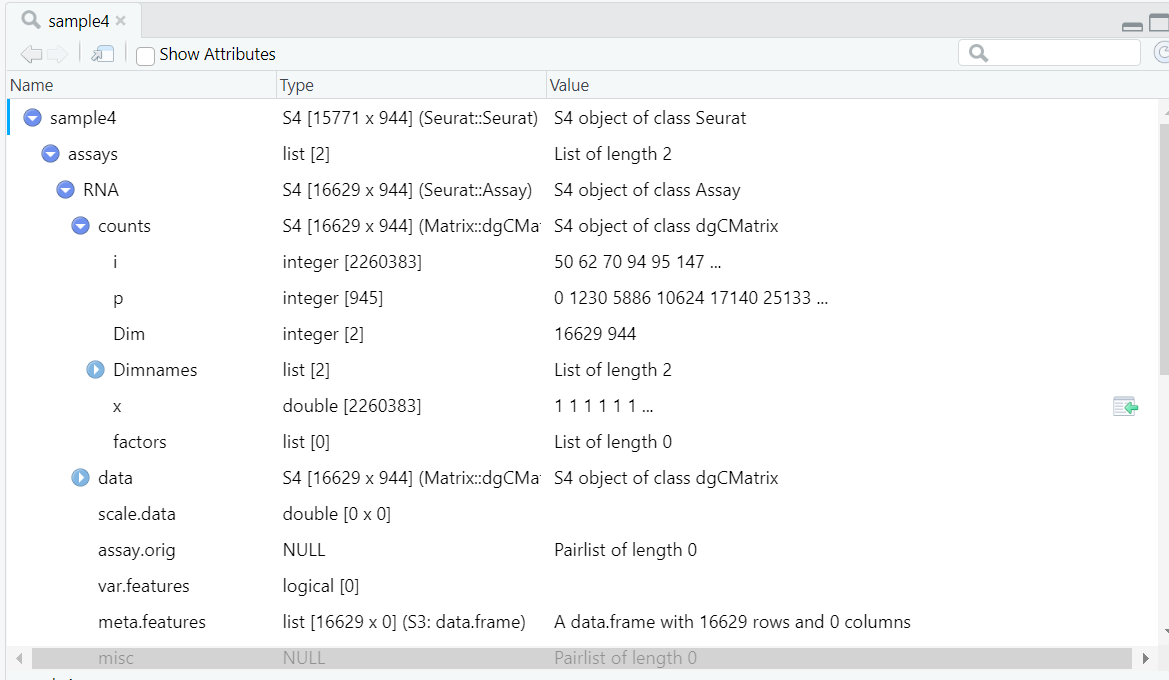 ~
~
# 导入包
library(Seurat)
library(scales)
save_dir = ‘/results’
fnm = paste0(save_dir,’/seurat_object.RData’)
load(fnm)
## 定义肿瘤区域
tumor_stroma_clu <- function(SID){
fnm = paste0(save_dir,’/MIA/A’,SID,’.xls’)
tmp1 = read.table(fnm,header = TRUE, stringsAsFactors = F, check.names=F)
tms = c(‘Ep_1′,’Ep_2′,’Ep_3′,’Ep_4’)
N_clu = dim(tmp1)[2]
TM_clu = c(‘100’) # tumor clusters
SM_clu = c(‘100’) # stroma clusters
for (i in seq(1,length(tms))){
for (j in seq(1,N_clu)){
if (tmp1[tms[i],j]>10){
TM_clu = c(TM_clu,colnames(tmp1)[j])
}
}
}
TM_clu = TM_clu[2:length(TM_clu)]
TM_clu = unique(TM_clu)
SM_clu = setdiff(colnames(tmp1),TM_clu)
return(list(TM_clu,SM_clu))
}
## 将所有肿瘤区域作为元数据汇集
pool_TM = function(SOE,TM_clu){
temp = SOE@meta.data$seurat_clusters
temp = as.character(temp)
for(i in seq(1,length(temp))){
if (temp[i] %in% TM_clu){
temp[i] = ‘tumor’
}
}
SOE@meta.data$pool_tumor = as.factor(temp)
return(SOE)
}
## 将所有基质区域作为元数据汇集
pool_SM = function(SOE,SM_clu){
temp = SOE@meta.data$seurat_clusters
temp = as.character(temp)
for(i in seq(1,length(temp))){
if (temp[i] %in% SM_clu){
temp[i] = ‘stroma’
}
}
SOE@meta.data$pool_stroma = as.factor(temp)
return(SOE)
}
## 定义变量
SID = ‘4’
SOE = sample4
TS = tumor_stroma_clu(SID)
TM_clu = unlist(TS[1])
SM_clu = unlist(TS[2])
SOE = pool_TM(SOE,TM_clu)
sample4 = pool_SM(SOE,SM_clu)
## 储存Seurat 项目
fnm = paste0(save_dir,’/seurat_object_TM_SM.RData’)
save(sample4,sample5,sample10,sample12, file = fnm) # Save the object
第二步:绘制肿瘤轮廓
在第五步的基础上,我们需要额外导入imager,magick,stringr这三个包,引用第五步中设置的肿瘤簇定位函数,进而绘制肿瘤簇轮廓,绘制的结果如下图所示,分别是四个样本的肿瘤簇轮廓。
# 导入包
library(magick)
library(stringr)
## 保存结果文件
folder = paste0(save_dir,’/map_tumor_region’)
if (!file.exists(folder)){
dir.create(folder)
}
folder1 = paste0(save_dir,’/map_tumor_outline’)
if (!file.exists(folder1)){
dir.create(folder1)
}
## 找到肿瘤簇
tum_clu <- function(SID){
fnm = paste0(save_dir,’/MIA/A’,SID,’.xls’)
tmp1 = read.table(fnm,header = TRUE, stringsAsFactors = F, check.names=F)
tms = c(‘Ep_1′,’Ep_2′,’Ep_3′,’Ep_4’)
# tms = c(‘T’)
# tms = c(‘FB_1′,’FB_2′,’FB_3’)
N_clu = dim(tmp1)[2]
TM_clu = c(‘100’)
for (i in seq(1,length(tms))){
for (j in seq(1,N_clu)){
if (tmp1[tms[i],j]>10){
TM_clu = c(TM_clu,colnames(tmp1)[j])
}
}
}
TM_clu = TM_clu[2:length(TM_clu)]
TM_clu = unique(TM_clu)
return(TM_clu)
}
## 绘制肿瘤区域
map_tumor <- function(SID,SOE,topleft,topright,bottomleft,bottomright,im,size = 7){
tumor_clu = tum_clu(SID)
tmp0 = SOE@meta.data$seurat_cluster
nspots = length(tmp0)
mycols1 <- c(rep(‘NA’,length(tmp0)))
for (i in seq(1,nspots)){
if (tmp0[i] %in% tumor_clu){
mycols1[i] = ‘black’
}
}
delta_x = mean(abs(topright[1]-topleft[1]),abs(bottomright[1]-bottomleft[1]))/32
delta_y = mean(abs(topright[2]-bottomright[2]),abs(topleft[2]-bottomleft[2]))/34
adjust_x = function(step){return((step-1)*(bottomleft[1]-topleft[1])/34)}
adjust_y = function(step){return((step-1)*(topright[2]-topleft[2])/32)}
tmp = as.matrix(GetAssayData(object = SOE, assay = “SCT”, slot = “scale.data”))
tmp1 = colnames(tmp)
if (nchar(SID)==1){
tmp1 = substr(tmp1,4,100)
} else{
tmp1 = substr(tmp1,5,100)
}
colnames(tmp) = tmp1
x = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][1]})))
y = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][2]})))
d = data.frame(x,y)
filenm = paste0(folder,’/A’,SID,’_tumor.png’)
png(filenm, width = dim(im)[1], height = dim(im)[2])
par(mar = c(0,0,0,0)) # set zero margins on all 4 sides
plot(x = NULL, y = NULL, xlim = c(0,dim(im)[1]), ylim = c(0,dim(im)[2]), pch = ”,
xaxt = ‘n’, yaxt = ‘n’, xlab = ”, ylab = ”, xaxs = ‘i’, yaxs = ‘i’,
bty = ‘n’) # plot empty figure
# rasterImage(im, xleft = 0, ybottom = 0, xright = dim(im)[1], ytop = dim(im)[2])
points(topleft[1]+(d[,1]-1)*delta_x+apply(d,1,function(x){return(adjust_x(x[2]))}),
topleft[2]-(d[,2]-1)*delta_y+apply(d,1,function(x){return(adjust_y(x[1]))}),
col=mycols1,pch=15,cex=size)
dev.off()
}
## 定义变量
SID = ‘4’
SOE = sample4
fnm = paste0(image_dir,’/Sample_’,SID,’_ds.jpg’) # downsample to 0.25x
im = load.image(fnm)
# plot(im)
xd = dim(im)[1]
yd = dim(im)[2]
topleft = c(58,yd-50)
topright = c(1412,yd-64)
bottomleft = c(48,yd-1512)
bottomright = c(1400,yd-1524)
map_tumor(SID,SOE,topleft,topright,bottomleft,bottomright,im,size=8.5)
*其余三个样本进行相同处理
## 绘制肿瘤轮廓
files1 = list.files(folder)
for (i in seq(1,length(files1))){
directory = paste0(folder,’/’,files1[i])
sample_id = str_split(files1[i],’.png’)[[1]][1]
fig <- image_read(directory)
# print(fig)
fig = fig %>% image_morphology(‘EdgeOut’, ‘Octagon’) %>% image_negate() # edge detection
filenm = paste0(folder1,’/’,sample_id,’_outline.png’)
image_write(fig, filenm)
}
A4
A5
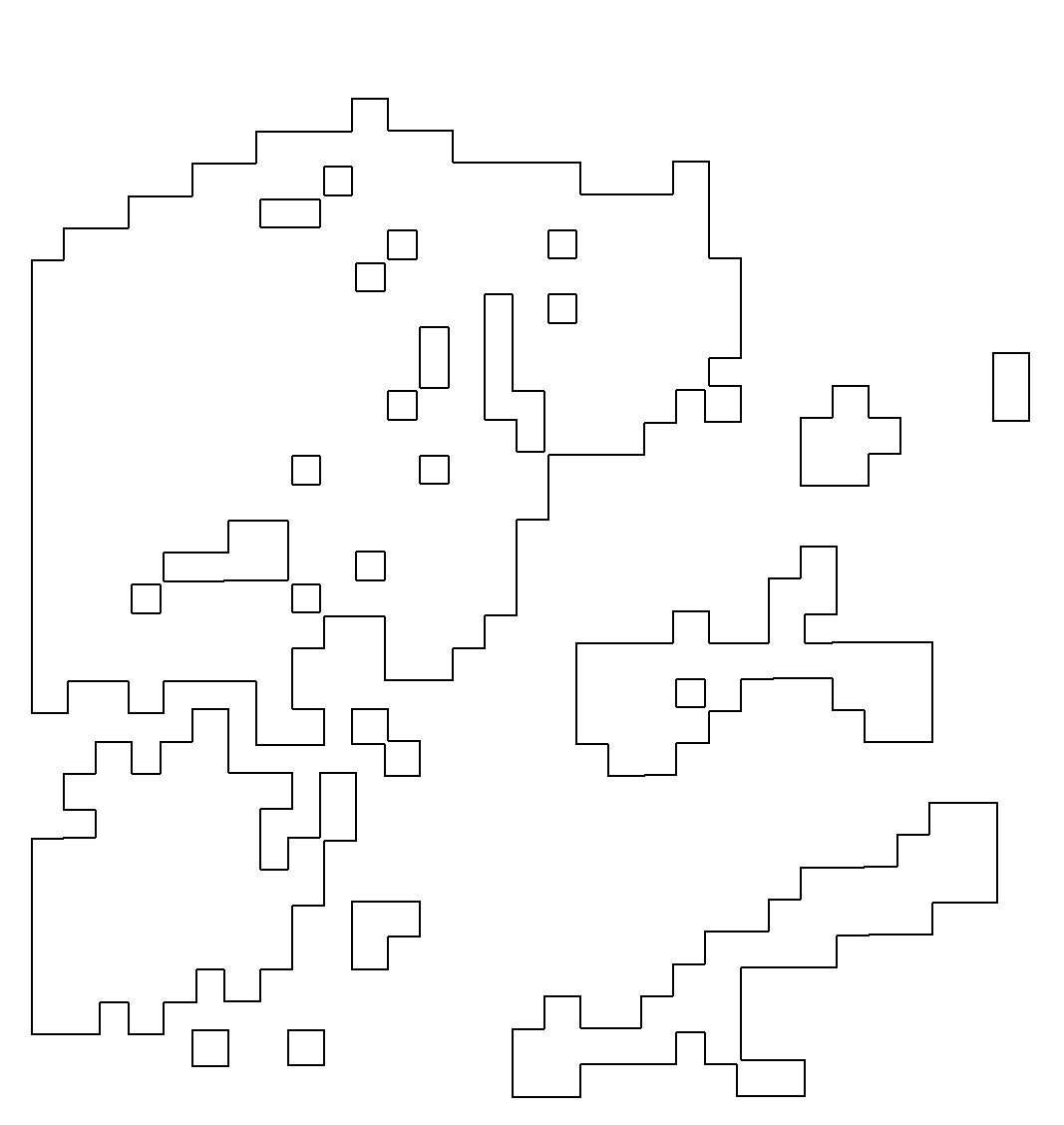
A10
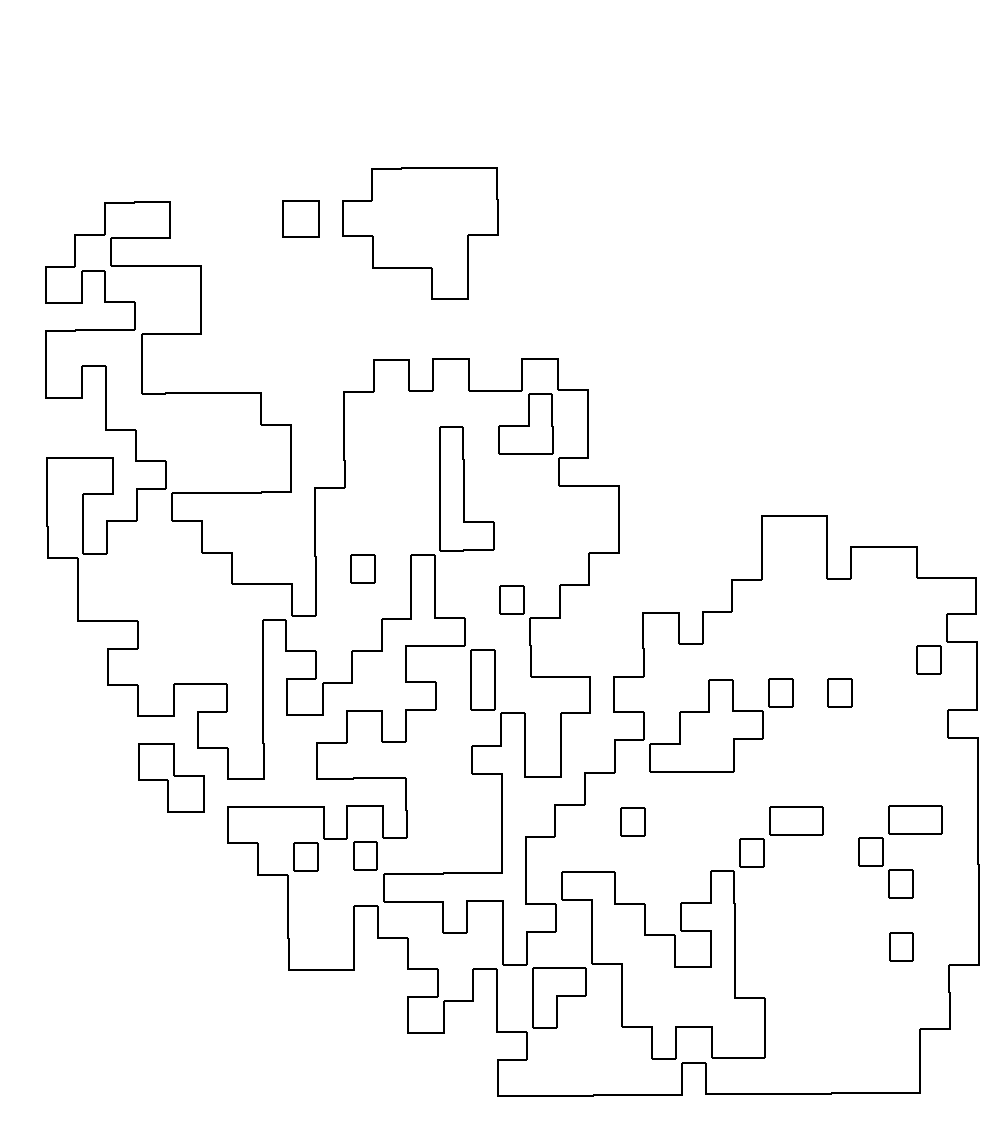
A12
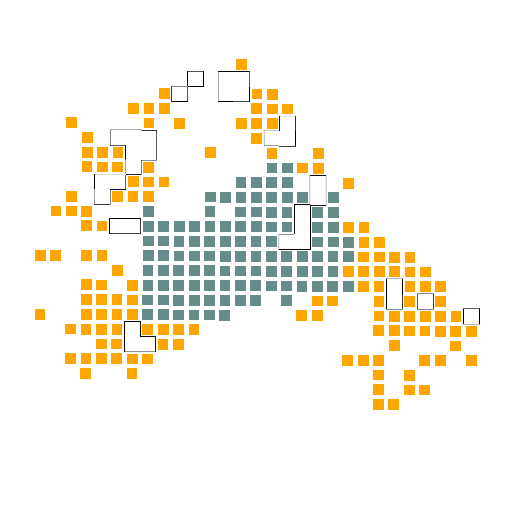
结果如图,是我们依据肿瘤的轮廓进行的绘图,可以非常清晰的看到肿瘤的界限,接着我们需要找出个体的基因,然后将其与肿瘤的轮廓进行比对。
第三步:与个体基因比对
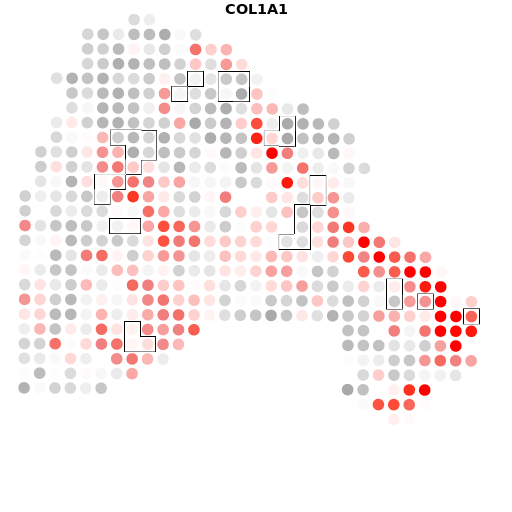
接下来,小果带着大家来找出scRNA中的特征基因,依据给出的代码,将肿瘤与基因相比对,就可以绘制出下方的图,当然,下方的四幅图只展示了COL1A1这个基因与四个样本的之间的映射关系,剩余的基因与肿瘤簇的映射关系小伙伴们可以自己在结果文件中找一找哦。
## 设定肿瘤,基质基因
geneb = c(‘POSTN’,’CD36′,’ACTA2′,’VIM’,’S100A4′,’PDGFRB’,’PDGFRA’,
‘FAP’,’COL1A1′,’WFDC2′,’MUC16′,’CLDN4′)
## 比对基因
map_gene <- function(gene,SID,SOE,RorG,topleft,topright,bottomleft,bottomright,im){
fnm = paste0(save_dir,’/map_tumor_outline/A’,SID,’_tumor_outline.png’) # downsample to 0.25x
im1 = load.image(fnm)
delta_x = mean(abs(topright[1]-topleft[1]),abs(bottomright[1]-bottomleft[1]))/32
delta_y = mean(abs(topright[2]-bottomright[2]),abs(topleft[2]-bottomleft[2]))/34
adjust_x = function(step){return((step-1)*(bottomleft[1]-topleft[1])/34)}
adjust_y = function(step){return((step-1)*(topright[2]-topleft[2])/32)}
tmp = as.matrix(GetAssayData(object = SOE, assay = “SCT”, slot = “scale.data”))
tmp1 = colnames(tmp)
if (nchar(SID)==1){
tmp1 = substr(tmp1,4,100)
} else{
tmp1 = substr(tmp1,5,100)
}
colnames(tmp) = tmp1
x = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][1]})))
y = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][2]})))
d = data.frame(x,y)
all_genes = rownames(tmp)
id = 0
for (i in seq(1,length(all_genes),by = 1)){
if (all_genes[i] == gene){
id = i
break}}
if (id == 0){
print(‘No matched genes!’)
}
data_1 = tmp[id,]
vec = (data_1-min(data_1))/(max(data_1)-min(data_1))
require(grDevices)
R = ‘brown’
G = ‘darkgreen’
B = ‘darkblue’
if (RorG==1){
mycols <- colorRamp(c(‘darkgray’,’white’,’lightcoral’,’red’), space = “Lab”)(vec)
} else if (RorG==2){
mycols <- colorRamp(c(“white”,G), space = “Lab”)(vec)
} else {
mycols <- colorRamp(c(“white”,B), space = “Lab”)(vec)
}
mycols <- rgb(mycols[,1], mycols[,2], mycols[,3], maxColorValue = 255) # Transform the rgb colors in hexadecimal format
filenm = paste0(folder,’/A’,SID,’_’,gene,’_with_tumor_gray.png’)
png(filenm, width = 512, height = 512)
par(mar = c(0,0,0,0)) # set zero margins on all 4 sides
plot(x = NULL, y = NULL, xlim = c(0,dim(im)[1]), ylim = c(0,dim(im)[2]), pch = ”,
xaxt = ‘n’, yaxt = ‘n’, xlab = ”, ylab = ”, xaxs = ‘i’, yaxs = ‘i’,
bty = ‘n’) # plot empty figure
rasterImage(im1, xleft = 0, ybottom = 0, xright = dim(im1)[1], ytop = dim(im1)[2])
points(topleft[1]+(d[,1]-1)*delta_x+apply(d,1,function(x){return(adjust_x(x[2]))}),
topleft[2]-(d[,2]-1)*delta_y+apply(d,1,function(x){return(adjust_y(x[1]))}),
col=mycols,pch=20,cex=3)
title(gene, line = -1)
dev.off()
# print(gene)
}
##
SID = ‘4’
SOE = sample4
fnm = paste0(image_dir,’/Sample_’,SID,’_ds.jpg’) # downsample to 0.25x
im = load.image(fnm)
# plot(im)
xd = dim(im)[1]
yd = dim(im)[2]
topleft = c(58,yd-50)
topright = c(1412,yd-64)
bottomleft = c(48,yd-1512)
bottomright = c(1400,yd-1524)
isRorG = 1
gene = geneb
genes = rownames(SOE@assays$SCT)
gene = intersect(gene,genes)
for (i in seq(1,length(gene))){
map_gene(gene[i],SID,SOE,isRorG,topleft,topright,bottomleft,bottomright,im)
}
## 绘制颜色条带
color.bar <- function(lut, min=0, max=1, nticks=11, ticks=seq(min, max, len=nticks), title=”) {
scale = (length(lut)-1)/(max-min)
filenm = paste0(folder,’/color_bar.png’)
png(filenm, width = 170, height = 500)
plot(c(0,10), c(min,max), type=’n’, bty=’n’, xaxt=’n’, xlab=”, yaxt=’n’, ylab=”, main=title)
axis(2, ticks, las=1)
for (i in 1:(length(lut)-1)) {
y = (i-1)/scale + min
rect(0,y,10,y+1/scale, col=lut[i], border=NA)
}
dev.off()
}
color.bar(colorRampPalette(c(‘darkgray’,’white’,’lightcoral’,’red’))(100))
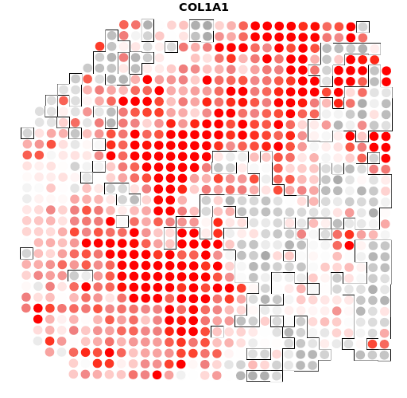
A4+COL1A1
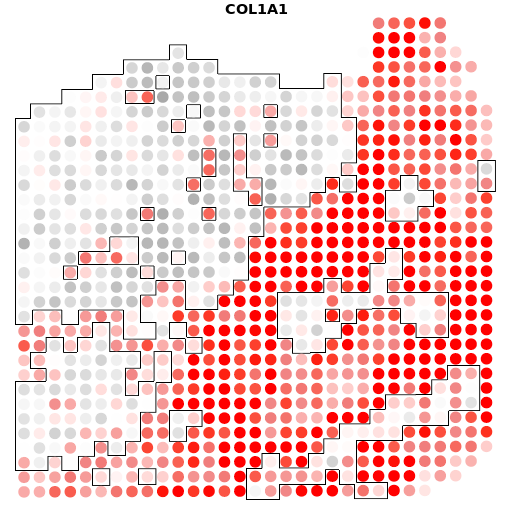
A10+COL1A1
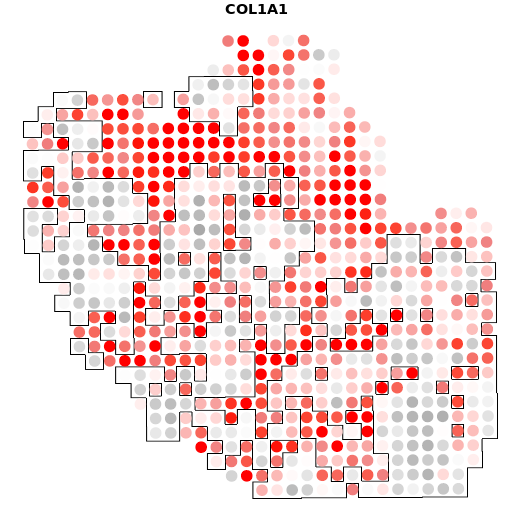
A12+COL1A1
A5+COL1A1
结果如图,是我们将个体基因与肿瘤的轮廓进行比对之后的结果,有很多的结果图,小果在这里只列出了一个基因的比对图,可以看出在四个样本中,COL1A1基因的表达是非常丰富的,图中的红色区域代表高表达的区域,灰色的则代表低表达的区域。
第四步:将选定的簇(例如CAF-癌症相关成纤维细胞)映射到具有肿瘤轮廓的图像上
只有肿瘤的轮廓是无法进行分析的,因此,必须要将特定的肿瘤簇绘制到肿瘤的轮廓上,我们需要导入额外的包:dplyr。然后按照代码将与癌症相关的成纤维细胞(CAF)比对至肿瘤轮廓上,在设置好四个样本的参数后,即可得到结果图。
## 将肿瘤的轮廓比对至癌症相关的成纤维细胞
map_clusters <- function(clusters,fib_clu,tum_clu,SID,SOE,topleft,topright,bottomleft,bottomright,im){
fnm = paste0(save_dir,’/map_tumor_outline/A’,SID,’_tumor_outline.png’)
im = load.image(fnm)
delta_x = mean(abs(topright[1]-topleft[1]),abs(bottomright[1]-bottomleft[1]))/32
delta_y = mean(abs(topright[2]-bottomright[2]),abs(topleft[2]-bottomleft[2]))/34
adjust_x = function(step){return((step-1)*(bottomleft[1]-topleft[1])/34)}
adjust_y = function(step){return((step-1)*(topright[2]-topleft[2])/32)}
tmp = SOE@meta.data$seurat_cluster
Nclu = length(levels(tmp))
nspots = length(tmp)
mycolpalette1 = c(‘orange’,’paleturquoise4′,’orchid’,’seagreen2′,’slateblue1′,’yellow’)
mycolpalette = c(‘red’,’darkgreen’,’blue’,’cyan’,’black’,’yellow’)
mycols <- c(rep(NA,length(tmp)))
for (i in seq(1,nspots)){
for (j in seq(1,length(fib_clu))){
if ((as.character(tmp[i]) == fib_clu[j]) && (as.character(tmp[i]) %in% clusters)){
mycols[i] = mycolpalette1[j]
}
}
}
filenm = paste0(save_dir,’/map_CAF_clusters’,’/A’,SID,’_CAF.png’)
png(filenm, width = 512, height = 512)
par(mar = c(0,0,0,0),’bg’=’lightgray’) # set zero margins on all 4 sides
plot(x = NULL, y = NULL, xlim = c(0,dim(im)[1]), ylim = c(0,dim(im)[2]), pch = ”,
xaxt = ‘n’, yaxt = ‘n’, xlab = ”, ylab = ”, xaxs = ‘i’, yaxs = ‘i’,
bty = ‘n’) # plot empty figure
rasterImage(im, xleft = 0, ybottom = 0, xright = dim(im)[1], ytop = dim(im)[2])
points(topleft[1]+(d[,1]-1)*delta_x+apply(d,1,function(x){return(adjust_x(x[2]))}),
topleft[2]-(d[,2]-1)*delta_y+apply(d,1,function(x){return(adjust_y(x[1]))}),
col=mycols,pch=15,cex=2)
dev.off()
}
##A4
SID = ‘4’
SOE = sample4
fnm = paste0(image_dir,’/Sample_’,SID,’_ds.jpg’)
im = load.image(fnm)
plot(im)
xd = dim(im)[1]
yd = dim(im)[2]
topleft = c(58,yd-50)
topright = c(1412,yd-64)
bottomleft = c(48,yd-1512)
bottomright = c(1400,yd-1524)
delta_x = mean(abs(topright[1]-topleft[1]),abs(bottomright[1]-bottomleft[1]))/32
delta_y = mean(abs(topright[2]-bottomright[2]),abs(topleft[2]-bottomleft[2]))/34
adjust_x = function(step){return((step-1)*(bottomleft[1]-topleft[1])/34)}
adjust_y = function(step){return((step-1)*(topright[2]-topleft[2])/32)}
tmp = as.matrix(GetAssayData(object = SOE, assay = “SCT”, slot = “data”))
tmp1 = colnames(tmp)
tmp1 = substr(tmp1,4,100)
colnames(tmp) = tmp1
x = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][1]})))
y = as.numeric(unlist(lapply(tmp1, function(x){strsplit(x,”x”)[[1]][2]})))
d = data.frame(x,y)
fib_clu = c(“0″,”1″,”3″,”4″,”6″,”7”)
tum_clu = c(“2″,”5”)
clusters = c(‘0′,’7’)
map_clusters(clusters,fib_clu,tum_clu,SID,SOE,topleft,topright,bottomleft,bottomright,im)
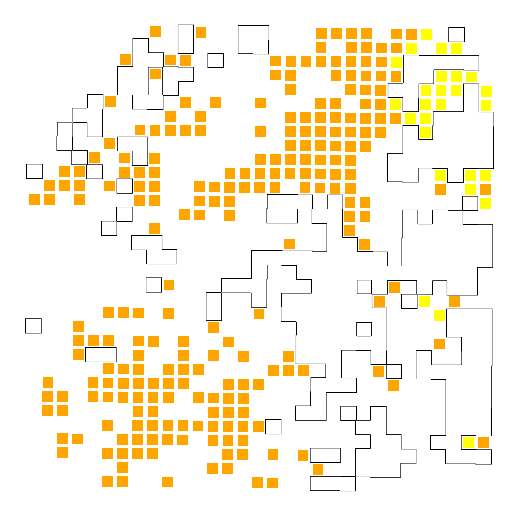
A4-CAF
A10-CAF
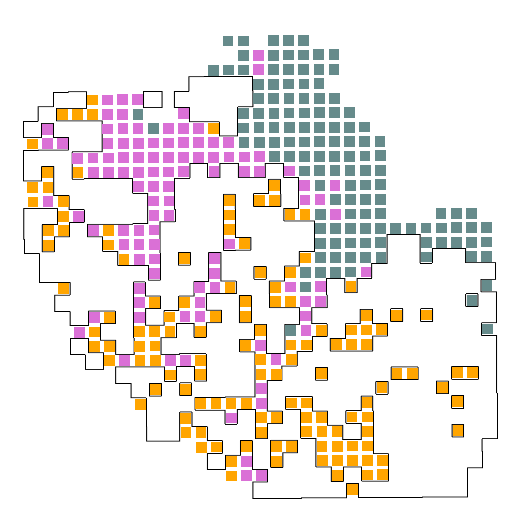
A5-CAF
A12-CAF
我们将自己选定的肿瘤簇(CAF)投射到之前绘出的轮廓图上,即可看到特定的簇在整个肿瘤轮廓中的分布,最后我们还需要对所研究的簇和整个背景簇进行基因的差异表达分析,看看究竟在特定的肿瘤簇中什么样的基因是表达最丰富的。
第五步:可视化簇logFC
在这一步,我们就要用到在文章一开始提到的avg_logFC了,不知道小伙伴们是否还记得呢?如果忘记的话赶快返回去看一下~
为了让我们的图片更加多彩,我们需要新导入两个R包:gplots,RcolorBrewer。然后提取不同簇中的最高和最低的差异化表达的基因,并记录他们的数量。接着提取出不同基因在不同样本中的差异表达程度,也就是平均对数折叠变化-avg_logFC,在这个基础上,根据平均对数折叠变化的相关性,对基因和样本进行聚类。最后,选择具有差异性的基因,进行可视化,就可以得到下面的两幅图。
# 提取出基因的差异表达信息
genes = c()
clusters = c()
for (i in seq(1,length(files))){
fnm = files[i]
clu = unlist(strsplit(fnm,’.x’))[1]
tmp1 = read.table(paste0(dir1,’/’,fnm),header = TRUE, stringsAsFactors = F, check.names=F)
tmp1 = tmp1[abs(tmp1$avg_logFC)>0.15,]
tmp1 = tmp1[tmp1$p_val<0.01,]
genes = c(genes,tmp1$gene[1:20])
temp = tmp1[order(tmp1$avg_logFC,decreasing = FALSE),]
genes = c(genes,temp$gene[1:20])
clusters= c(clusters,clu)
}
genes = unique(genes)
df = data.frame(matrix(0, ncol = length(clusters), nrow = length(genes)))
row.names(df) = genes
colnames(df) = clusters
for (i in seq(1,length(files))){
fnm = files[i]
tmp1 = read.table(paste0(dir1,’/’,fnm),header = TRUE, stringsAsFactors = F, check.names=F)
for (j in seq(1,length(genes))){
if (genes[j] %in% tmp1$gene){
df[genes[j],i] = tmp1$avg_logFC[tmp1$gene==genes[j]]
}
}
}
my_palette <- colorRampPalette(brewer.pal(10, “RdBu”))(256)
data = data.matrix(df)
## 根据最远的两个元素之间的距离来合并不同的簇
hr <- hclust(as.dist(1-cor(t(data), method=”spearman”)), method=”complete”)
hc <- hclust(as.dist(1-cor(data, method=”spearman”)), method=”complete”)
data[data<(-1)] = -1
data[data>1] = 1
filename = paste0(folder,’/avg_logFC_heatmap_spearman.pdf’)
pdf(filename,12,12)
heatmap.2(data,
col = rev(my_palette),
Rowv=as.dendrogram(hr), Colv=as.dendrogram(hc),
trace=”none”,
scale = “none”,
density.info=”none”,
margins=c(5,6),
lhei=c(1,8), lwid=c(1,4),
keysize=0.2, key.par = list(cex=0.5),
cexRow=0.9,cexCol=1.2,srtCol=90, # rotate column label
key.title = ‘avg_logFC’,
key.xlab = ‘avg_logFC’
)
dev.off()
## 为CAF绘制可视化的选择的基因
genes = c(‘ACTA2′,’S100A4′,’FAP’,’VIM’,’PDGFRB’,’PDGFRA’,’CD36′,’POSTN’,’COL1A1′)
CAF_clusters = c(“A4_c0”, “A4_c1”, “A4_c3”, “A4_c4”, “A4_c6”, “A4_c7”,
“A5_c0”, “A5_c1”, “A5_c2”, “A5_c3”, “A5_c4”, “A5_c6”,
“A10_c0”, “A10_c1”, “A10_c4”,
“A12_c2”, “A12_c3”, “A12_c4”)
df = data.frame(matrix(0, ncol = length(CAF_clusters), nrow = length(genes)))
row.names(df) = genes
colnames(df) = CAF_clusters
for (i in seq(1,length(CAF_clusters))){
fnm = paste0(dir1,’/’,CAF_clusters[i],’.xls’)
tmp1 = read.table(fnm, header = TRUE, stringsAsFactors = F, check.names=F)
for (j in seq(1,length(genes))){
if (genes[j] %in% tmp1$gene){
if (tmp1$p_val[tmp1$gene==genes[j]]<0.01){
df[genes[j],i] = tmp1$avg_logFC[tmp1$gene==genes[j]]
}
}
}
}
my_palette <- colorRampPalette(brewer.pal(10, “RdBu”))(256)
data = data.matrix(df)
data[data<(-1)] = -1
data[data>1] = 1
filename = paste0(folder,’/avg_logFC_CAF_genes.pdf’)
pdf(filename,11,11)
heatmap.2(data,
col = rev(my_palette),
dendrogram = ‘row’,
Colv= FALSE,
Rowv= TRUE,
distfun = dist,
hclustfun = hclust,
trace=”none”,
scale = “none”,
density.info=”none”,
margins=c(5,7),
lhei=c(1,6), lwid=c(1,4),
keysize=0.4, key.par = list(cex=0.5),
cexRow=1.1,cexCol=1,srtCol=90, # rotate column label
key.title = ‘avg_logFC’,
key.xlab = ‘avg_logFC’
)
dev.off()
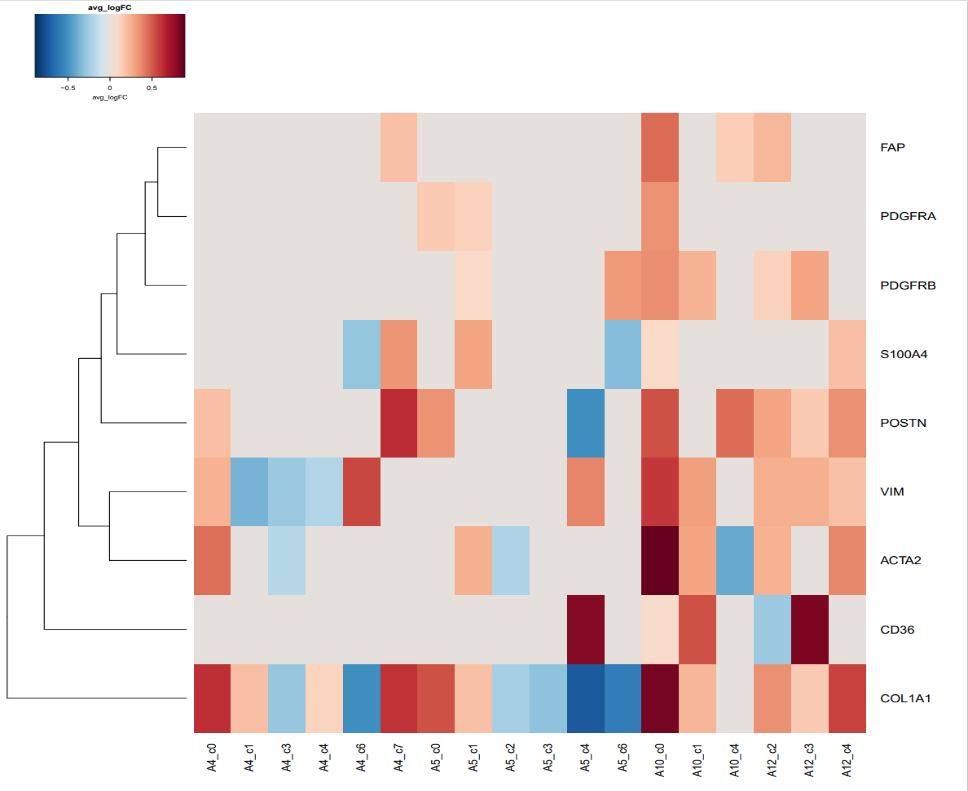
avg-logFC-CAF
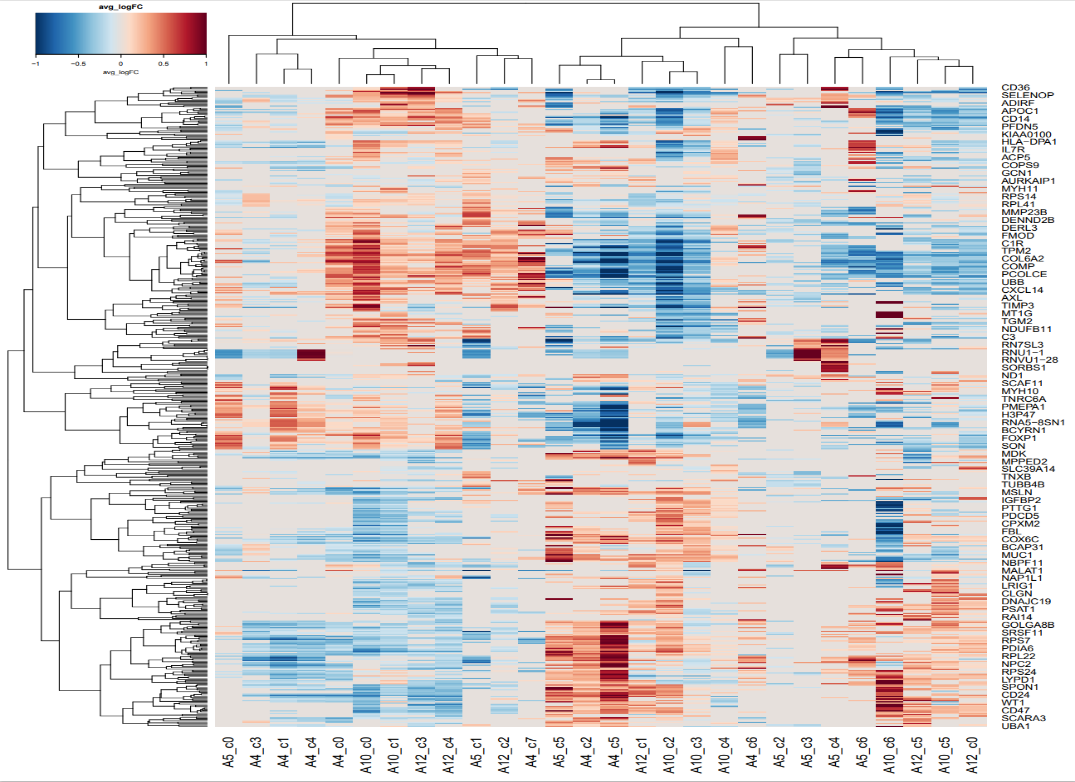
avg-logFC-heatmap
这两幅图中的第一幅图表示了众多的基因与四个样本中的不同区域的差异性,可以发现有很多表达并不具有明显的差异性的基因,因此我们需要在此基础之上进行选择,将最具有表达差异性的基因挑选出来,于是就有了第二幅图,从图中我们可以看到,COL1A1在四个样本之间的差异性最大,图中左上角的呃颜色条带表明,颜色越红,正值越大,在前一组的表达越明显,颜色越蓝,负值越大,在后一组的表达越明显,可以看到COL1A1这个基因的差异性还是非常的大的。
小果的分享就到这里啦,今天我们利用Seurat包以及一些相关的拓展包完成了初步的肿瘤差异化分析,主要是对特定肿瘤的基因差异进行了分析,从而找到了在肿瘤簇中具有较高表的的基因,这对于后续的靶向分析是非常重要的。
如果小伙伴们在绘图的过程中有任何疑问,可以试一试我们的云生信小工具哦,只要输入合适的数据以及指令就可以直接绘制想要的图呢,链接:http://www.biocloudservice.com/home.html。