各位小伙伴大家好,小果又和大家见面了,今天想和大家聊一下群体遗传相关内容。群体遗传学是遗传学的一个分支,研究的是群体水平上基因的传递和变异。它对理解物种的演化、种群的遗传结构以及遗传变异的维持和传递具有重要的意义,不仅能判断每个小群体的个数,还能对群体间的基因交流情况及物种的起源进行判断。
目前群体遗传常用的软件有strcture、admixture,这两个软件均能对进源祖先的群体结构进行分析,但两相比较之下strcture运行时间要慢一些,因此admixture可能更受欢迎。但作为老牌群体结构分析软件,它在行业内依然占据重要地位。目前strcture分别支持Linux、windows以及Mac版本,因为涉及到格式转换问题,所以我更推荐Linux系统下的structure,这可以减少各位小伙伴切换软件的频率,减少不必要的麻烦。但Linux系统终究也有弱点,就是不能及时出图,因此需要配合第三方软件R来进行图形绘制。当然啦,Linux及win中都有R,也就是说你要是学会了以下代码内容,各位小伙伴也是可以直接在Linux中实现图形绘制的哦!
我们通常以堆叠柱状图的形式展示群体结构结果,pophelper包就能实现该诉求,且还有更多的函数等待大家的开发。但是本次介绍的R包使用时较为耗费系统资源,并非很建议利用电脑运行,希望大家使用服务器运行,如果没有自己的服务器欢迎联系我们进行服务器租赁哦~
下面让我们一起来学习这个厉害的R包吧!!!
#R包的安装(1)
install.packages(‘devtools’)
devtools::install(‘royfrancis/pophelper’)
如果出现以下报错,可以使用第二种安装方法

#R包的安装(2)
install.packages(c(‘ggplot2′,’gridExtra’,’label’,’switching’,’tidyr’,’remotes’),
repos = “http://cloud.r-project.org”)
#检查是否成功安装,如果成功安装可报pophelper的版本信息
library(pophelper)

下面让我们开始真正使用该R包:
#首先设置工作目录,我的structure结果文件被存储在‘F:\群体遗传学习\2.群体结构分析\2.5structure分析’中
setwd(‘F:\\群体遗传学习\\2.群体结构分析\\2.5structure分析’)
#读取文件,小伙伴们在做structure时可知,其输入文件如下,因此为避免重复书写无用代码,我们利用循环进行输入
files <- list.files(path = ‘.’, #文件路径
pattern = ‘_f$’, #以_f结尾的文件
full.names = F) #是否展示路径信息
files #查看files的内容
#Q矩阵阅读
slist <- readQ(files = files, #文件
indlabfromfile = T, #文件中每行的命名
filetype = ‘structure’) #文件类型auto’, ‘structure’,’tess2′,’baps’,’basic’ or ‘clumpp’
#对齐
xlist <- alignK(slist)
#合并,共生成11个列表形式的文件,对应一开始读入的11个k值文件
xlist2 <- mergeQ(qlist)
#如果报错如下

#需要利用以下函数解决,可直接复制,但正常输出的结果都可用上述mergeQ进行合并,应为alignK继而mergeQ是同一个函数下的功能且应用顺序紧挨着
mergy <- function(x) {
return(list(Reduce(`+`, x)/length(x)))
}
fun1 <- function(x) as.matrix(unlist(attributes(x)))
a <- lapply(xlist, fun1)
b <- as.data.frame(t(as.data.frame(lapply(a, function(x,y) x[y, ], “k”), stringAsFactors = FALSE)), stringsAsFactors = FALSE)
fun2 <- function(x) if (all(!is.na(as.numeric(as.character(x))))) {
return(as.numeric(as.character(x)))
}else {
return(x)
}
b <- as.data.frame(sapply(b, fun2), stringAsFactors = FALSE)
ord <- order(b[, “k”])
qlist <- xlist[ord]
labels <- summariseQ(tabulateQ(qlist, sorttable = FALSE))$k
x <- unlist(lapply(splitQ(qlist), mergy), recursive = FALSE)
names(x) <- labels
xlist2 <- as.qlist(x)
xlist2
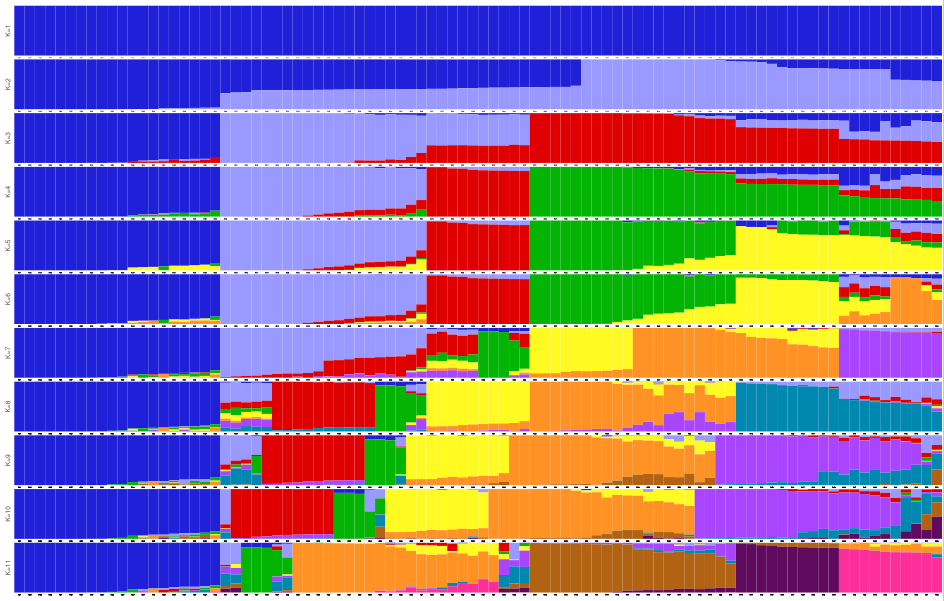
#随后进行图形绘制
P1 <- plotQ(xlist2, #Q矩阵
#图片合并,因为我们输入了11个文件所以不能选用sep(单个输出)
imgoutput = “join”,
imgtype = “pdf”,#输出格式”png”,”jpeg”,”tiff” or “pdf”.
width = 20,height = 1.2, #宽度和高度
showindlab = T, #是否对每个K值图形绘制边框
useindlab = T, #展示单株标签
indlabhjust = 0.5, #单株标签水平居中
indlabangle = 0, #单株标签角度
indlabsize = 1.2, #单株标签文本大小
sortind = ‘all’, #排序,基于聚类排序
#K值图不会共享单株标签名,且只在 imgoutput = “join”时有效
sharedindlab = F,
panelspacer = 0.02, #不同k值图的间隙
indlabspacer = 0, #不同k值图中单株空隙
indlabcol = “black”, #k值文本颜色
showtitle = T,titlelab = “”,titlecol = “black”,titlehjust = 0.5, #标题
showsp = T,sppos = “left”, #k值标签展示,也有‘right’
splab = paste0(“K=”,1:11), #k值标签
splabangle = 180, #标签角度
dpi = 600, #输出分辨率
outputfilename = paste0(“structure1”), #输出文件的名称
units = “cm”, #输出文件的大小
exportplot = T, #图片保存
exportpath = getwd()) #保存路径
#届时会在自己设置的工作目录下出现名为structure1.pdf的目标文件
#此外我们还可以进行分组显示
#引入分组信息
pop <- read.table(‘group.txt’, header = T, sep = ‘\t’, stringsAsFactors = F,row.names = 1)
pop
##绘图
P2 <- plotQ(xlist2,imgoutput = “join”,
imgtype = “png”, #输出格式的变化
height = 1.2,width = 20,
showindlab = T,useindlab = T,sharedindlab = T,
showsp = T,sppos = “left”,splab = paste0(“K=”,1:11),splabangle = 180,
showlegend = F,ordergrp = T,grplab = pop, #添加分组
subsetgrp = LETTERS[1:9], #添加组名
dpi = 600,
outputfilename = paste0(“structure_barplot_pop1”),units = “cm”,
exportpath = getwd())
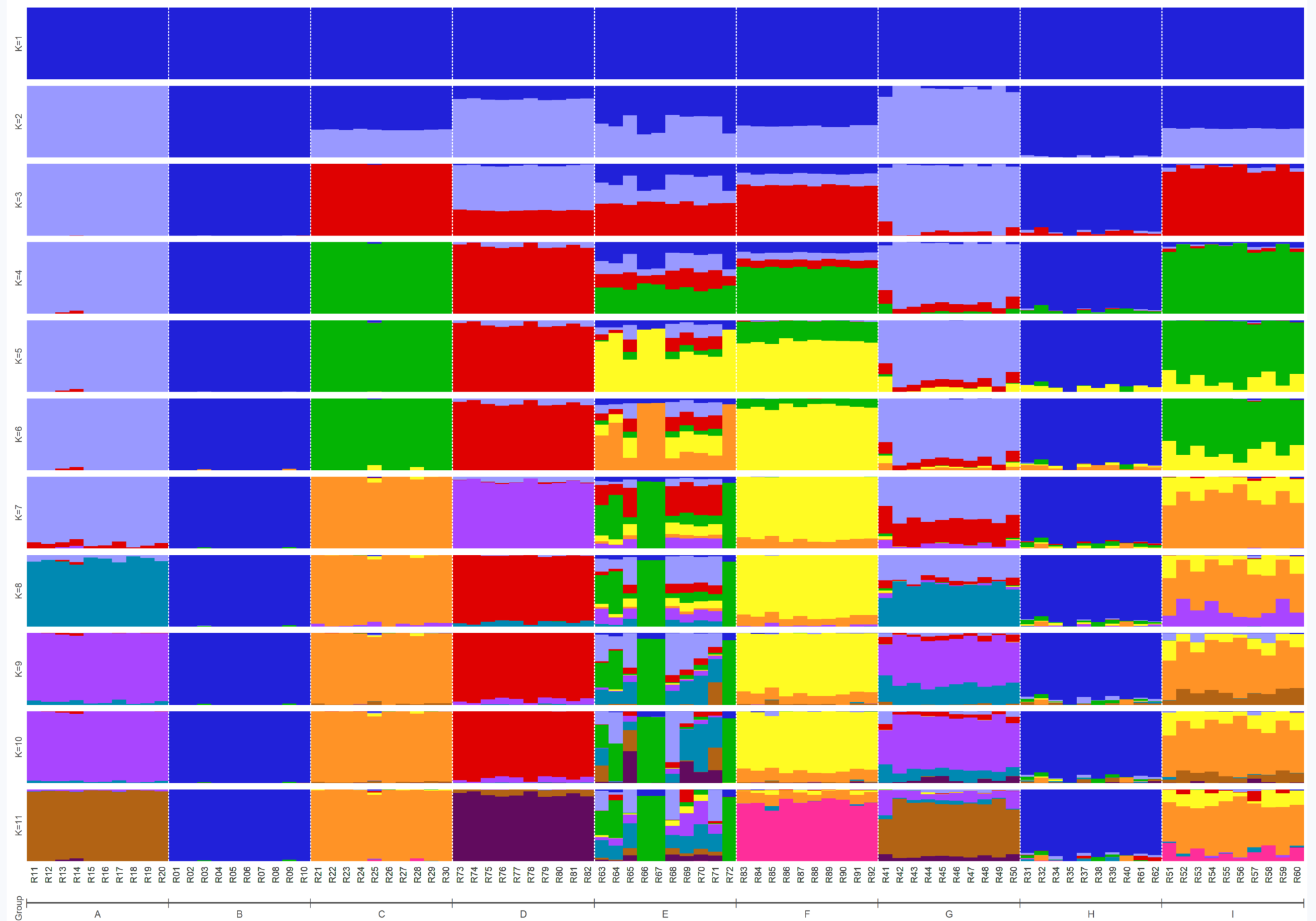
#此时已经可以在结构图下面看到分组信息
#最后推断最优的分群数目
#利用k值及deltaK靓列数确定最优k值,deltaK最大的最优
dt <- evannoMethodStructure(summariseQ(tabulateQ(slist,writetable = F,sorttable = T)),writetable = T,exportpath = getwd())
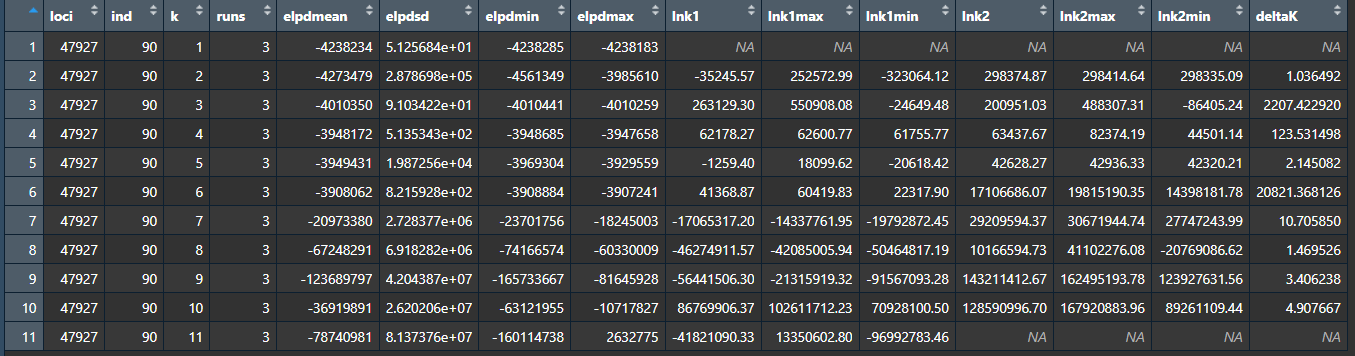
#写出最优Q矩阵,第6个是最优的
write.table(x = xlist2[[6]],file =’best_k6_Qscore’,quote = F,sep = ‘\t’)
那既然大家已经看到了这里,相信你们也是完美的进行了复现,那咱们现在就对结果图进行详细的解释吧!
咱们把带有分组信息的P2当做最后的结果图,从下往上看呢,首先可以看到A-I的9个分组,而在这9个分组的范围之内均有以R开头的菌株存在,其实这是小果自己对样品进行的分组哦,大家如果想要改变的话也是可以的啊。这个分组可以根据大家的实验来决定哦,比如不同的采样时间、不同的采样地点或同一属的不同种样品都是可以的。
然后大家继续向上看,可以发现共有11个长方形的堆叠柱形图,这对应着在一开始时输入的11个分类信息,即分别假设这90个样品分为1、2、3、……、11个群体进行计算,每个计算3次,以此计算deltaK值,所获得的deltaK值越大,说明该值对应的K即种群数越可信,如以上数据显示当K=6时deltaK达到了20000+,远远高于其余组的deltaK值,因此小果可以认为这是最优的分群数。
在此情况之下,我们将目光上下移动分别查看K为5和7时的分群状况。当K=5时,与K=6相比在E、G、H中有较大变化,即出现了以橙色为代表的一个小种群,既然K=6才是最有情况,则可以说明在这三个小群体之间,已经发生了生殖隔离等条件使得新种群正在逐渐形成。而当K=7时,除去颜色影响,其实并没有太大的变化。即这个图主要是展示某个群体、某一些样品是否为一类样品,或看某个样品是从哪里进化而来。
以上所写内容就是本期小果带大家一起学习的pophelper包的全部内容啦,其实该包内容极其强大,函数数量远不仅如此,如果小伙伴们觉得还想进行学习的话,就请赶快加入我们把!!该函数及代码的确繁复复杂,如果各位小伙伴觉得数据有问题或是代码运行不起来,可以试试小果自己的云生信小程序哦,里面有对多种数据进行处理及可视化的成熟代码,只需要动动手指就能直接出图,而且还接受颜色、字体等个性化定制哦,有需要的小伙伴可以点击下方链接吧!!
http://www.biocloudservice.com/home.html