数据库是生信分析的强大数据基础,往期的文章中小果给小伙伴们介绍了许多的数据库使用的方法,那么有没有一种方法可以同时分析多个数据库的数据呢?嘿嘿,小果连夜学习了可以同时对多个数据库的肿瘤数据进行免疫浸润的分析方法,如果小伙伴们感兴趣,就跟着小果一起来学习吧!考虑到本次复现的数据量比较大,小果推荐大家租赁我们的服务器进行本次的复现学习~
本次我们使用的是下面这几个数据库:
TARGET:https://ocg.cancer.gov/programs/target
CBTN:https://cbtn.org/research/specimendata/
ICGC :https://icgc.org/icgc/
TCGA:https://www.cancer.gov/about-nci/organization
在这几个数据库中,我们今天主要使用其中的儿童患者数据为大家进行教程,小伙伴们如果对其他的肿瘤数据感兴趣,也可以自己试一试其他的数据呦,我们在数据库中获取儿科神经系统肿瘤(pedNST)的RNA-seq数据,小果在这里已经帮大家整理好了,大家从下面的链接就可以获取:
链接:https://pan.baidu.com/s/135jKVqVbFB8CuAvDAGmgFw
提取码:9if2
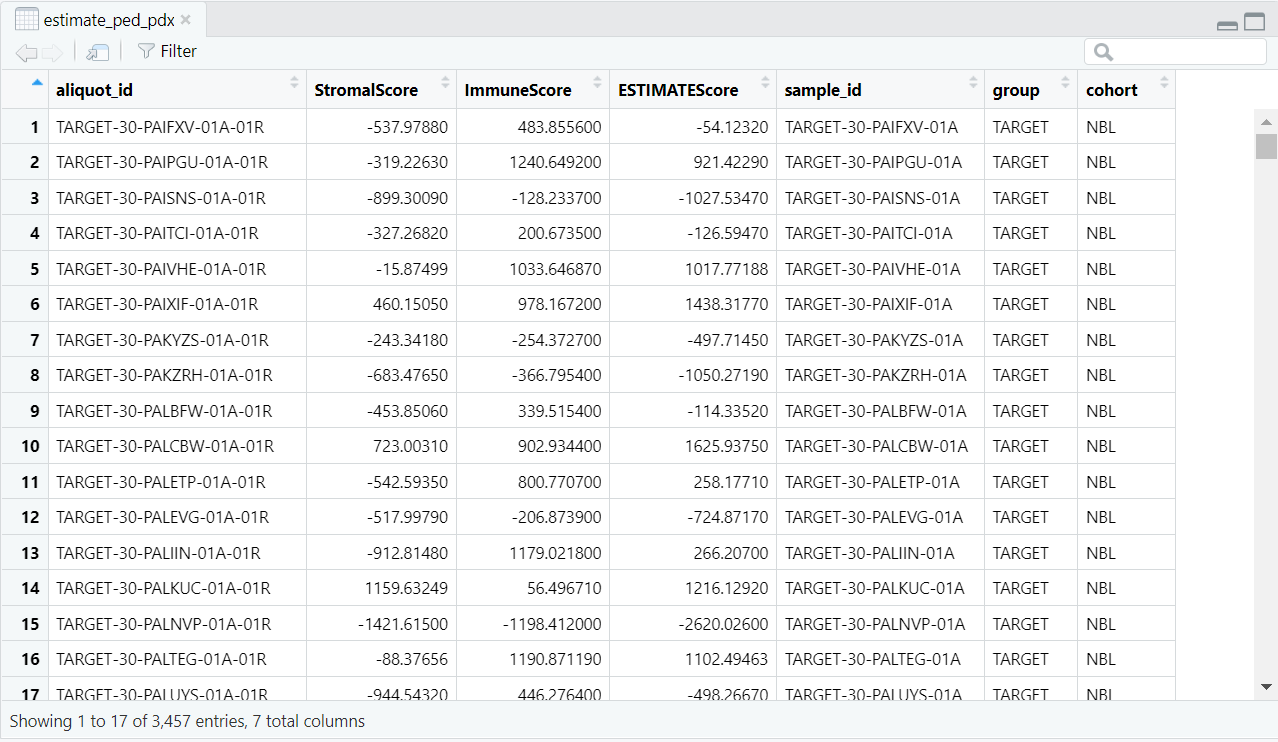
如图,是我们本次要用到的主要数据(estimate_ped_pdx.RData),其中包括了儿童的一些肿瘤数据,例如StromalScore,是一种基于基因表达数据预测肿瘤组织中基质细胞浸润程度的分值,数值越高,预后活性越差,并且免疫活性越低,还有免疫得分,ESTIMATE免疫评分等。
在分析之前,小果先给大家介绍一下今天的分析思路,我们先导入下载的数据,然后依次对数据进行总体的概览,利用一些评分标准进行评分,划分免疫集群,最后进行一些相关性分析,这个思路同样可以应用到其他的免疫分析中~
Step 1 对导入的四个数据库的样本量和研究的肿瘤类型进行概览
#导入整合好的数据库数据
load(paste0(datapath,”estimate_ped_pdx.RData”))
#只保留ped(儿科)数据
ped <- estimate_ped_pdx[ estimate_ped_pdx$group != “TCGA”,]
tab <- as.data.frame(table(ped$cohort), stringsAsFactors = F)
tab <- tab[order(tab$Freq, decreasing = T),]
#排序
ped$cohort <- factor(ped$cohort, levels = tab$Var1)
ped$group <- factor(ped$group, levels = c(“CBTN”, “ICGC”, “TARGET”, “PDX (ITCC)”))
fig1a <- ggplot(data = ped) + geom_bar(aes(y = cohort, fill = group)) + myaxis + myplot +
theme(axis.title = element_blank(), axis.text.x = element_text(size = 25, angle = 0, hjust = 0.5),
legend.position = c(0.8,0.9), legend.title = element_blank(), plot.margin = margin(1,1,1,1, “cm”)) +
scale_fill_manual(values = group_col)
pdf(file = paste0(plotpath,”Fig1_A.pdf”),
width = 10, height = 10, useDingbats = FALSE)
fig1a
dev.off()
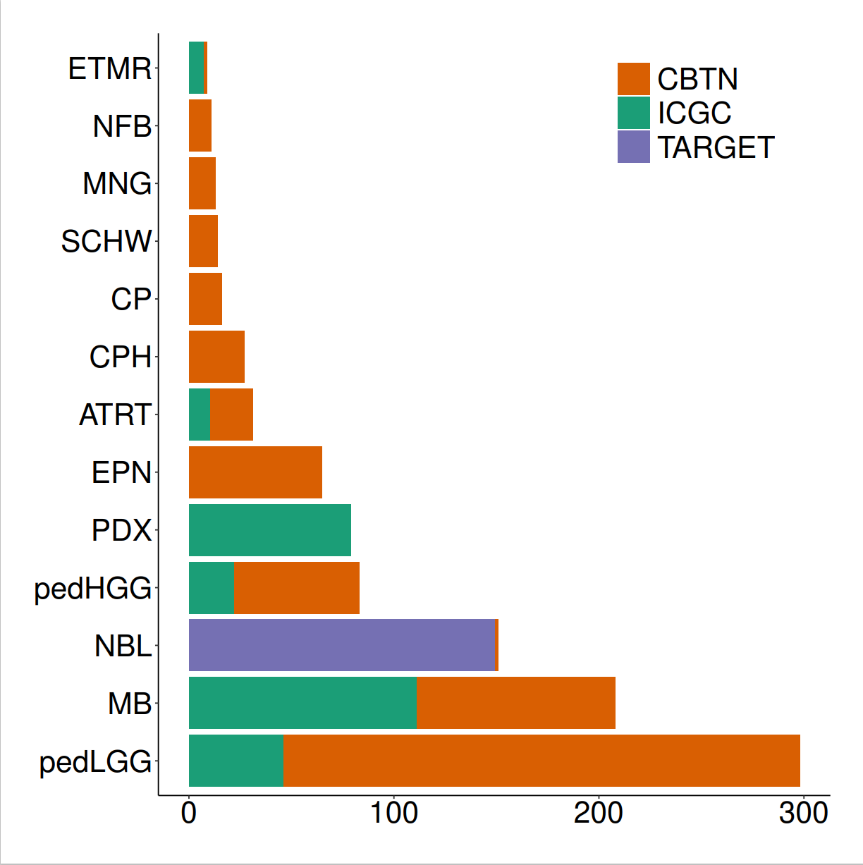
如图是概览的结果,图中显示了不同肿瘤类型(如ETMR、NFB等)在CBTN、ICGC和TARGET三个数据库中的数量。其中,PDX:患者来源的异种移植模型,ETMR:带有多层花簇的胚胎性肿瘤,NFB:神经纤维瘤,MNG:脑膜瘤,SCHW:神经鞘瘤,CP:脉络丛肿瘤,CPH:颅咽管瘤,ATRT:非典型畸胎瘤/横纹肌样肿瘤,EPN:室管膜瘤,pedHGG:儿科高级别胶质瘤,NBL:神经母细胞瘤,MB:髓母细胞瘤,pedLGG:儿科低级别胶质瘤。说明其中的肿瘤类型还是非常丰富的。
Step 2 对所有的肿瘤数据进行ESTIMATE免疫评分分析
#设置数组
emptyvar <- as.data.frame(matrix(ncol = 7, nrow = 2))
colnames(emptyvar) <- colnames(estimate_ped_pdx)
emptyvar$group <- as.character(emptyvar$group)
emptyvar$cohort <- as.character(emptyvar$cohort)
emptyvar[1,”aliquot_id”] <- “empty1”
emptyvar[2,”aliquot_id”] <- “empty2”
emptyvar[1,”sample_id”] <- “empty1”
emptyvar[2,”sample_id”] <- “empty2”
emptyvar[1,”ImmuneScore”] <- 3500
emptyvar[2,”ImmuneScore”] <- 3500
emptyvar[1,”cohort”] <- “EMPTY1”
emptyvar[2,”cohort”] <- “EMPTY2”
estimate_ped_pdx <- rbind(estimate_ped_pdx,emptyvar)
#计算ESTIMATE免疫评分
estimate_ped_pdx$percread <- 8.0947988*exp(estimate_ped_pdx$ImmuneScore*0.0006267)
immune.cohorts <- cbind(NA, unique(estimate_ped_pdx$cohort))
colnames(immune.cohorts) <- c(“group”,”cohort”)
immune.cohorts <- as.data.frame(immune.cohorts)
immune.cohorts$group <- as.character(immune.cohorts$group)
adults <- c(“PRAD”, “LGG”, “OV”, “SKCM”, “COAD”, “GBM”, “LUAD”)
peds <- c(“PDX”,”ETMR”, “MB”, “ATRT”, “EPN”, “pedHGG”, “CP”, “NBL”, “pedLGG”, “CPH”, “MNG”, “SCHW”, “NFB”)
immune.cohorts[immune.cohorts$cohort %in% adults, 1] <- “Adult”
immune.cohorts[immune.cohorts$cohort %in% peds, 1] <- “Pediatric”
immune.cohorts[immune.cohorts$cohort == “EMPTY1”,1] <- “Pediatric”
immune.cohorts[immune.cohorts$cohort == “EMPTY2”,1] <- “Pediatric”
#获取每一组的药物数据
for(i in 1:nrow(immune.cohorts)){
immune.cohorts$median_immunereads[i]<-median(estimate_ped_pdx$percread[estimate_ped_pdx$cohort == immune.cohorts$cohort[i]])
}
#对肿瘤的类型进行排序
tmp <- immune.cohorts[which(immune.cohorts$group == “Pediatric”),]
tmp1 <- immune.cohorts[which(immune.cohorts$group == “Adult”),]
immune.cohorts <- rbind(tmp,tmp1)
immune.cohorts$cohort <- factor(immune.cohorts$cohort, levels = c(“PDX”,”ETMR”, “MB”, “ATRT”, “EPN”, “pedHGG”, “CP”,
“NBL”, “pedLGG”, “CPH”, “MNG”, “SCHW”, “NFB”,
“EMPTY1″,”EMPTY2”, “PRAD”, “LGG”, “OV”, “SKCM”,
“COAD”, “GBM”, “LUAD”))
immune.cohorts <- immune.cohorts[order(immune.cohorts$cohort),]
# 调用Splot 对数据进行处理,准备绘图
disease.width <- (nrow(estimate_ped_pdx)/nrow(immune.cohorts))
sorted.estimate_ped_pdx <- estimate_ped_pdx[0,]
start = 0
for(i in 1:(nrow(immune.cohorts))){
tmp <- estimate_ped_pdx[estimate_ped_pdx$cohort == immune.cohorts$cohort[i],]
tmp <- tmp[order(tmp$percread),]
#create range of x values to squeeze dots into equal widths of the plot for each Disease regardless of the number of samples
div <- disease.width/nrow(tmp)
#If there is only one sample, put the dot in the middle of the alloted space
if(dim(tmp)[1]==1)
{
tmp$Xpos<-start+(disease.width/2)
} else tmp$Xpos<-seq(from = start, to = start+disease.width, by = div)[-1]
sorted.estimate_ped_pdx<-rbind(sorted.estimate_ped_pdx,tmp)
immune.cohorts$Median.start[i] <- tmp$Xpos[1]
immune.cohorts$Median.stop[i] <- tmp$Xpos[nrow(tmp)]
immune.cohorts$N[i]<-nrow(tmp)
start <- start+disease.width+30
}
immune.cohorts$medianloc <- immune.cohorts$Median.start+((immune.cohorts$Median.stop-immune.cohorts$Median.start)/2)
sorted.estimate_ped_pdx$cohort <- factor(sorted.estimate_ped_pdx$cohort, levels = levels(immune.cohorts$cohort))
#颜色设置
rmEMPTY <- rep(“black”,22)
rmEMPTY[14:15] <- “white”
immune.cohorts$color_crossbar <- NA
immune.cohorts$color_crossbar[immune.cohorts$cohort == “EMPTY1”] <- “white”
immune.cohorts$color_crossbar[immune.cohorts$cohort == “EMPTY2”] <- “white”
immune.cohorts$color_crossbar[is.na(immune.cohorts$color_crossbar)] <- “black”
immune.cohorts$cohort_n <- paste0(immune.cohorts$cohort, ” (n=”, immune.cohorts$N, “)”)
#绘制图片
fig1b1.fx <- function(x){
ggplot() +
geom_point(data = sorted.estimate_ped_pdx, aes(x = Xpos ,y = percread, color = cohort),
size = 7, shape = 20) +
geom_crossbar(data = immune.cohorts,
aes(x = medianloc,y = median_immunereads, color = color_crossbar,
ymin = median_immunereads,ymax = median_immunereads), width = disease.width) +
myaxis + myplot +
theme(axis.title.x = element_blank(), axis.title.y = element_text(size = 25),
axis.text.x = element_text(size = 25, angle = 45, hjust = 1, color = rmEMPTY),
axis.text.y = element_text(size = 25)) +
scale_color_manual(values = c(cohort_col, “white” = “white”, “black” = “black”), guide = “none”) +
scale_x_continuous(breaks = seq((disease.width)/2,max(sorted.estimate_ped_pdx$Xpos),
disease.width+30), labels = immune.cohorts$cohort_n, expand = c(0,20)) +
scale_y_continuous(breaks = seq(0, 70, by = 10)) +
labs(y = “% Immune Reads”)
}
fig1b <- fig1b1.fx(sorted.estimate_ped_pdx)
pdf(file = paste0(plotpath,”Fig1_B.pdf”),
width = 20, height = 8, useDingbats = FALSE)
fig1b
dev.off()
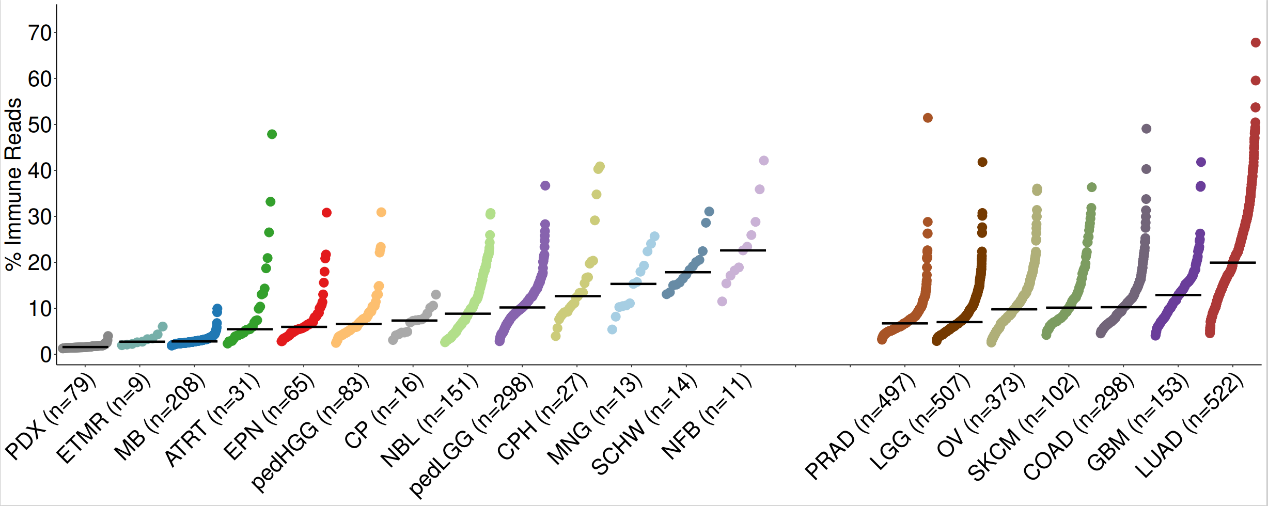
如图,是基于ESTIMATE免疫评分的儿童和成人癌症的百分比免疫读数分布。ESTIMATE,也就是Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data,是一种使用基因表达数据预测肿瘤纯度和肿瘤组织中浸润性基质/免疫细胞存在的分数,包含基质评分(用于捕捉肿瘤组织中基质的存在) ,免疫评分(用于表示肿瘤组织中免疫细胞的浸润) ,estimate评分(用于推断肿瘤纯度)。
图中的PDX是作为免疫浸润的阴性对照而设置的,每种颜色代表一种特定类型的癌症,y轴表示%免疫读数,x轴上列出了各种癌症类型及其样本数量。
通过ESTIMATE免疫评分可以映了免疫系统对肿瘤的反应强度。从图中可以看出,不同类型的癌症在免疫读数上有很大的差异,肾细胞癌、黑色素瘤和卵巢癌等癌症的免疫读数也较高。这对于研究癌症的免疫治疗和免疫耐受是有重要意义的。
Step 3 对不同的免疫集群进行划分
#加载数据
load(file = paste0(datapath,”metadata_IC.RData”))
load(file = paste0(datapath, “geneset_cc_normalized.RData”))
#基于免疫簇进行排序
cluster_cohort <- metadata_IC[order(metadata_IC$immune_cluster, metadata_IC$cohort),]
#提取免疫簇信息
mycluster <- as.character(cluster_cohort$immune_cluster)
names(mycluster) <- rownames(cluster_cohort)
cluster_hm <- class_hm.fx(mycluster)
#提取肿瘤类型的信息
mycohort <- cluster_cohort$cohort
names(mycohort) <- rownames(cluster_cohort)
mycohorts <- t(as.matrix(mycohort))
rownames(mycohorts) <- “Cohort”
cohorts_hm <- cohorts_hm.fx(mycohorts)
cells_mat <- geneset_cc_norm[,rownames(cluster_cohort)]
# 绘制热图
cells_hm <- cells_hm.fx(cells_mat)
#进行注释
annotation_order <- c(“Pediatric Inflammed”, “Myeloid Predominant”, “Immune Neutral”, “Immune Desert”)
cluster_ha = HeatmapAnnotation(clusters = anno_mark(at = c(50, 235, 566, 844), labels_rot = 0,
labels = annotation_order, side = “top”,
labels_gp = gpar(fontsize = 20),
link_height = unit(0.5, “cm”)))
fig1c <- cluster_ha %v% cluster_hm %v% cells_hm %v% cohorts_hm
#设置参数
lgd_cohort = Legend(labels = names(cohort_col)[2:13], title = “”, nrow = 1, legend_gp = gpar(fill = cohort_col[2:13]))
pdf(paste0(plotpath,”Fig1_C.pdf”),
width = 18, height = 10)
draw(fig1c, annotation_legend_side = “bottom”, legend_grouping = “original”,
annotation_legend_list = list(lgd_cohort))
dev.off()
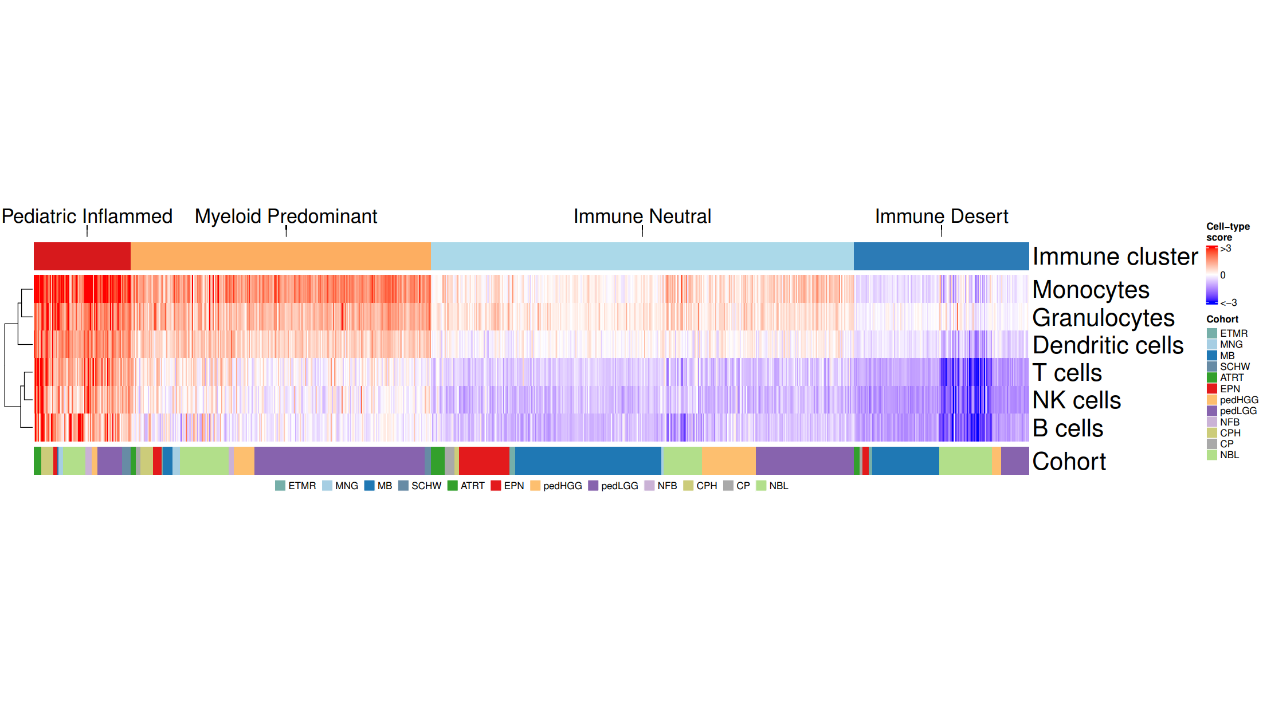
分析的结果如图所示,图片的顶部有四个类别,分别是Pediatric Inflamed(炎症型),Myeloid Predominant(髓系主导型),Immune Neutral(免疫中性型),和Immune Desert(免疫沙漠型),是根据免疫细胞的总体水平或比例进行划分的。
而图片的右侧有一个图例,显示了不同颜色代表的免疫细胞类型,例如单核细胞,粒细胞,树突状细胞,T细胞,NK细胞,和B细胞。这些免疫细胞都是人体免疫系统的重要组成部分,参与抵抗感染和肿瘤等病理过程。
在图片的底部有一些彩色的条形,用来表示不同的肿瘤类型,中间部分是一个矩阵,每个小方格中的颜色用来表示某一种免疫细胞在某一类别或队列中的丰度,颜色越深表示丰度越高,颜色越浅表示丰度越低。从矩阵中可以看出,不同类别或队列之间的免疫细胞分布和丰度有很大的差异,例如,Pediatric Inflamed类别中的NK细胞和树突状细胞丰度较高,而Immune Desert类别中的免疫细胞丰度普遍较低,这些差异可能反映了不同条件下的免疫状态和功能。
Step 4 癌症与特定免疫集群的相关性分析
#导入数据
load(file = paste0(datapath,”metadata_IC.RData”))
tab <- as.data.frame(table(metadata_IC$cohort), stringsAsFactors = F)
tab <- tab[order(tab$Freq, decreasing = F),]
# 将肿瘤簇和肿瘤类型汇聚为矩阵
cancer_IC_mat <- matrix(nrow = 12, ncol = 4,
dimnames = list(tab$Var1,
c(“Pediatric Inflamed”, “Myeloid Predominant”, “Immune Neutral”, “Immune Desert”)))
for(i in 1:nrow(cancer_IC_mat)){
mycancer <- metadata_IC[ metadata_IC$cohort == rownames(cancer_IC_mat)[i],]
freq_tab <- as.data.frame(table(mycancer$immune_cluster), stringsAsFactors = F)
freq_tab$perc <- freq_tab$Freq/sum(freq_tab$Freq)
cancer_IC_mat[i, freq_tab$Var1] <- freq_tab$perc *100
}
#处理NA数据
cancer_IC_mat[is.na(cancer_IC_mat)] <- 0
# 定义簇
row_dend <- as.dendrogram(hclust(dist(cancer_IC_mat), “complete”))
row_dend <- dendextend::rotate(row_dend,
c(“NFB”, “SCHW”, “MNG”, “CPH”, “ATRT”, “NBL”, “pedLGG”, “ETMR”, “MB”, “CP”, “pedHGG”, “EPN”))
# 注释
ha = rowAnnotation(`cohort size` = anno_barplot(tab$Freq, bar_width = 1,
gp = gpar(col = “white”, fill = “#4d4d4d”),
border = FALSE,
axis_param = list(at = c(0, 100, 200, 300), labels_rot = 45, gp = gpar(fontsize = 20)),
width = unit(3, “cm”)),
show_annotation_name = FALSE)
ha_1 = HeatmapAnnotation(`immune size` = anno_barplot( as.matrix(table(metadata_IC$immune_cluster)), bar_width = 1,
gp = gpar(col = “white”, fill = “#4d4d4d”),
border = FALSE,
axis_param = list(at = c(0, 100,200,300), labels_rot = 45, gp = gpar(fontsize = 20)),
height = unit(3, “cm”)),
show_annotation_name = FALSE)
# 绘图
col_fun= colorRamp2(c(0, 100), c(“white”, “red”))
cancer_hm = Heatmap(cancer_IC_mat,
#titles and names
name = “% cancer”,
show_row_names = TRUE,
show_column_names = TRUE,
#clusters and orders
cluster_columns = FALSE,
cluster_rows = row_dend,
show_row_dend = TRUE,
#aesthestics
row_names_side = “left”,
col = col_fun,
column_names_rot = 45,
column_names_gp = gpar(fontsize = 10),
row_names_gp = gpar(fontsize = 10),
height = unit(nrow(cancer_IC_mat), “cm”),
width = unit(ncol(cancer_IC_mat), “cm”),
column_title_gp = gpar(fontsize = 10),
column_title = NULL,
row_title = NULL,
right_annotation = ha,
top_annotation = ha_1,
show_heatmap_legend = TRUE)
pdf(paste0(plotpath,”Fig1_D.pdf”),
width = 10, height = 10)
draw(cancer_hm)
dev.off()
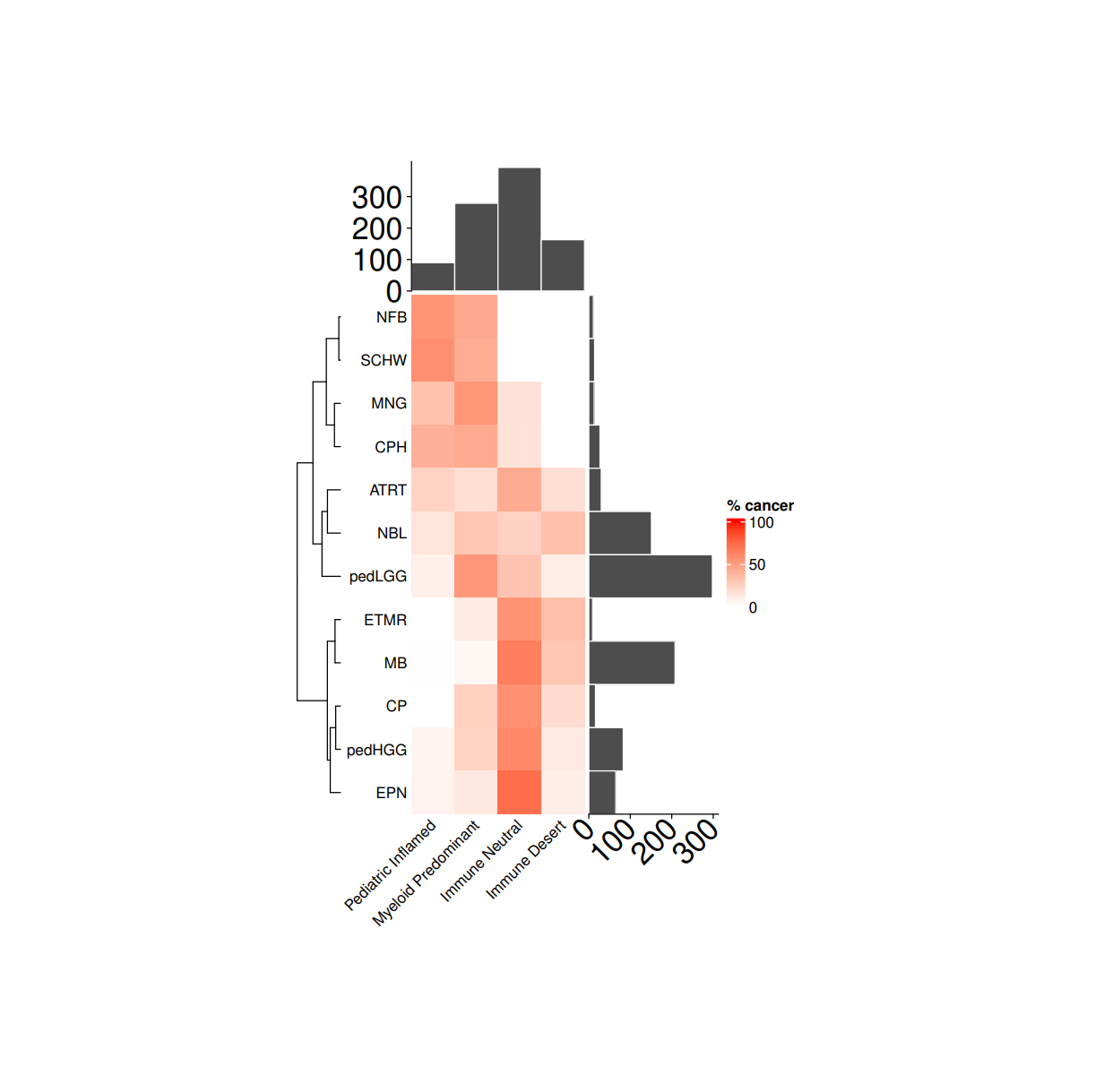
结果如图,显示了不同类型的癌症与特定免疫集群的表达量之间的关系。图中纵轴列出了不同类型的肿瘤(NFB, SCHW, JMNG 等),横轴列出了免疫集群,同时,在纵轴上还有一个渐变色条,表示0到100%的癌症发生率。(癌症发生率是指某一基因在某一类别中的样本中表达异常的比例,反映了该肿瘤与免疫集群的相关性)。
每个单元格的颜色代表该肿瘤在特定集群中的癌症发生率;颜色越深,百分比越高,表示该基因与该类别更加相关;颜色越浅,百分比越低,表示该基因与该类别不太相关。例如,NFB基因在ImmuDesert中的癌症发生率为0%,而EPN在Immune Neutral类别中的癌症发生率为100%,说明EPN与Immune Neutral类别有很强的相关性,可能是EPN的致病因素或者诊断指标。
Step 5 分析免疫类型与CRI-iAtlas簇(免疫集群)的关系
#导入数据
load(file = paste0(datapath,”metadata_IC.RData”))
tab <- as.data.frame(table(metadata_IC$CRI_cluster), stringsAsFactors = F)
tab <- tab[order(tab$Freq, decreasing = F),]
#存储每个免疫集群和免疫簇的百分比
cri_IC_mat <- matrix(nrow = 6, ncol = 4,
dimnames = list(tab$Var1,c(“Pediatric Inflamed”, “Myeloid Predominant”,
“Immune Neutral”, “Immune Desert”)))
for(i in 1:nrow(cri_IC_mat)){
mycancer <- metadata_IC[ metadata_IC$CRI_cluster == rownames(cri_IC_mat)[i],]
freq_tab <- as.data.frame(table(mycancer$immune_cluster), stringsAsFactors = F)
freq_tab$perc <- freq_tab$Freq/sum(freq_tab$Freq)
cri_IC_mat[i, freq_tab$Var1] <- freq_tab$perc *100
}
cri_IC_mat[is.na(cri_IC_mat)] <- 0
col_fun= colorRamp2(c(0, 100), c(“white”, “red”))
#设置热图参数
cri_hm = Heatmap(cri_IC_mat,
#titles and names
name = “% CRI-iAtlas cluster”,
show_row_names = TRUE,
show_column_names = TRUE,
#clusters and orders
cluster_columns = FALSE,
cluster_rows = FALSE,
show_column_dend = TRUE,
#aesthestics
row_names_side = “left”,
col = col_fun,
column_names_rot = 45,
column_names_gp = gpar(fontsize = 15),
row_names_gp = gpar(fontsize = 15),
height = unit(nrow(cri_IC_mat), “cm”),
width = unit(ncol(cri_IC_mat), “cm”),
column_title_gp = gpar(fontsize = 15),
column_title = NULL,
row_title = NULL,
show_heatmap_legend = TRUE)
ha = rowAnnotation(
`cohort size` = anno_barplot(tab$Freq, bar_width = 1,
gp = gpar(col = “white”, fill = “#4d4d4d”),
border = FALSE,
axis_param = list(gp = gpar(fontsize=15), at = c(0, 100,250,500), labels_rot = 45),
width = unit(4, “cm”)),
show_annotation_name = FALSE)
pdf(paste0(plotpath, “Fig1_E.pdf”), width = 10, height = 10)
cri_hm + ha
dev.off()
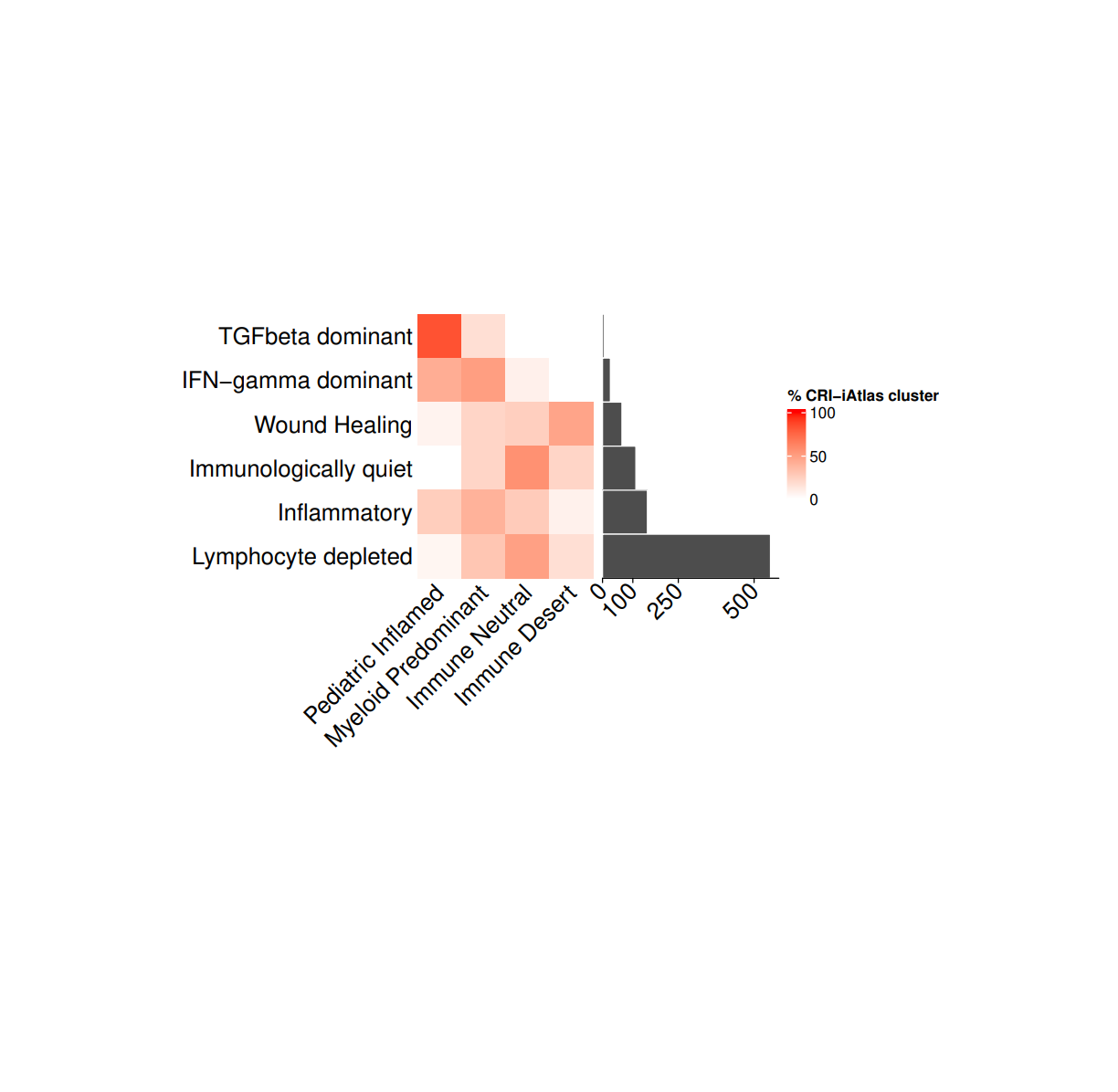
分析的结果如图,左边的热图显示了pedNST中每个CRI-iAtlas簇(免疫集群)中跨越免疫簇的样本比例,而柱状图显示了每个CRI-iAtlas簇聚类的样本总数,从而展示了不同免疫反应类型与CRI-iAtlas簇的数量之间的关系。
例如:Y轴列出了七种不同的免疫反应类型:TGFbeta dominant(TGF-β占优势), IFN-gamma dominant(IFN-γ占优势), Wound Healing(创伤愈合), Immunologically quiet(免疫静默), Inflammatory(炎症), Lymphocyte depleted(淋巴细胞耗竭)和Mixed(混合型),表中用红色、白色来表示每种免疫反应类型对应的CRI-iAtlas簇的数量。红色代表较高数量,白色代表较低数量。
Step 6 用H&E TIL分数分析免疫系统对肿瘤的反应强度
#导入数据(HE评分)
load(file = file.path(datapath,”HE_manifest.RData”))
#绘图
heplot <- ggplot(data = HE_manifest,
aes(x = immune_cluster, y = agg_tilScore)) +
geom_beeswarm(cex = 1.5, aes(color = cohort), size = 5) +
geom_boxplot(width = 0.5, outlier.colour = NA, fill = NA) +
myaxis + myplot +
scale_color_manual(values = cohort_col) +
theme(legend.position = “none”,
plot.margin = unit(c(0.2,0.2,0.2,2), “cm”),
axis.title.x = element_blank(),
axis.title.y = element_text(size = 30),
axis.text.x = element_text(size = 30),
axis.text.y = element_text(size = 30),
plot.title = element_text(size = 30, hjust = 0.5)) +
geom_signif(comparisons = list(c(“Pediatric Inflamed”, “Myeloid Predominant”)), y_position = 0.4,
map_signif_level=TRUE, textsize = 10, test = “wilcox.test”, vjust = 0.5) +
geom_signif(comparisons = list(c(“Pediatric Inflamed”, “Immune Desert”)), y_position = 0.45,
map_signif_level=TRUE, textsize = 10, test = “wilcox.test”, vjust = 0.5) +
labs(y = “Average TIL score”) + ggtitle(~underline(“H&E TIL score (n = 355)”))
pdf(paste0(plotpath,”Fig1_F.pdf”),
width = 10, height = 12)
print(heplot)
dev.off()
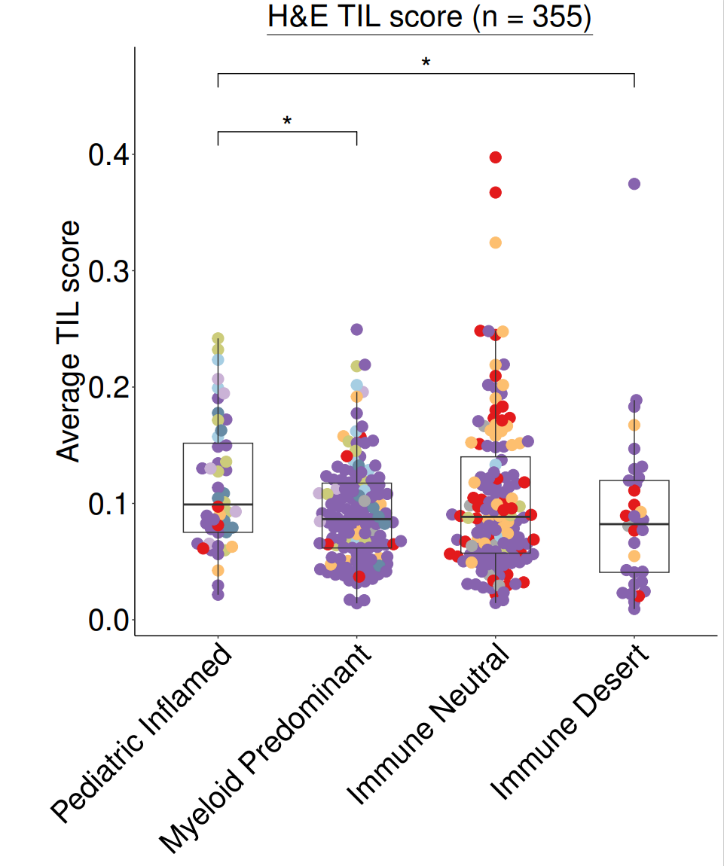
结果如图,我们在这一步中绘制的热图是一个显示H&E TIL分数和免疫集群关系的散点图。
对于H&E TIL分数,这是一种用来评估肿瘤浸润性淋巴细胞(TIL)的方法,基于H&E染色的肿瘤切片,用于反映了免疫系统对肿瘤的反应强度。
图中不同颜色和形状的点表示不同疾病或样本来源,例如CBTN,ICGC,TCGA。每个集群中点的位置表示每个样本的H&E TIL分数,越靠近y轴表示分数越高,越靠近x轴表示分数越低。这里用箱形图来显示TIL评分,通过对整个儿科中枢神经系统肿瘤样本(CBTN)的病理图像进行分割分析而确定。所示的样本图像分别代表1%(下)、10%(中)和15%(上)的TIL分数,分别对应于低四分位数、平均值和高四分位数。
除此之外图中还有一些显著性标记,用来显示不同类别之间的H&E TIL分数是否有显著差异,例如,Pediatric Inflamed和Myeloid Predominant之间有一个星号,表示它们之间的差异是显著的,而Pediatric Inflamed和Immune Desert之间有一个星号,同样表示它们之间的差异是非常显著的。
Step 7 肿瘤与信号通路频率分析
#导入数据
load(file = paste0(datapath,”metadata_IC.RData”))
n_x <- 4
#
lggp_c <- subgroupcount_IC.fx(metadata_IC, “pedLGG”)
lggp_f <- subgroupfreq_IC.fx(metadata_IC, “pedLGG”)
hggp_c <- subgroupcount_IC.fx(metadata_IC, “pedHGG”)
hggp_f <- subgroupfreq_IC.fx(metadata_IC, “pedHGG”)
nblp_c <- subgroupcount_IC.fx(metadata_IC, “NBL”)
nblp_f <- subgroupfreq_IC.fx(metadata_IC, “NBL”)
atrtp_c <- subgroupcount_IC.fx(metadata_IC, “ATRT”)
atrtp_f <- subgroupfreq_IC.fx(metadata_IC, “ATRT”)
mbp_c <- subgroupcount_IC.fx(metadata_IC, “MB”)
mbp_f <- subgroupfreq_IC.fx(metadata_IC, “MB”)
epnp_c <- subgroupcount_IC.fx(metadata_IC, “EPN”)
epnp_f <- subgroupfreq_IC.fx(metadata_IC, “EPN”)
#组合条形图
c_ls = list(atrtp_c + ggtitle(expression(~underline(“ATRT”))),
nblp_c+ ggtitle(expression(~underline(“NBL”))),
mbp_c+ ggtitle(expression(~underline(“MB”))),
hggp_c + ggtitle(expression(~underline(“pedHGG”))),
lggp_c + ggtitle(expression(~underline(“pedLGG”))),
epnp_c + ggtitle(expression(~underline(“EPN”))))
f_ls = list(atrtp_f, nblp_f, mbp_f, hggp_f, lggp_f, epnp_f )
#合并
pdf(paste0(plotpath, “Fig1_G.pdf”),
width = 20, height = 10, useDingbats = FALSE)
wrap_plots(c(c_ls, f_ls), nrow = 2)
dev.off()
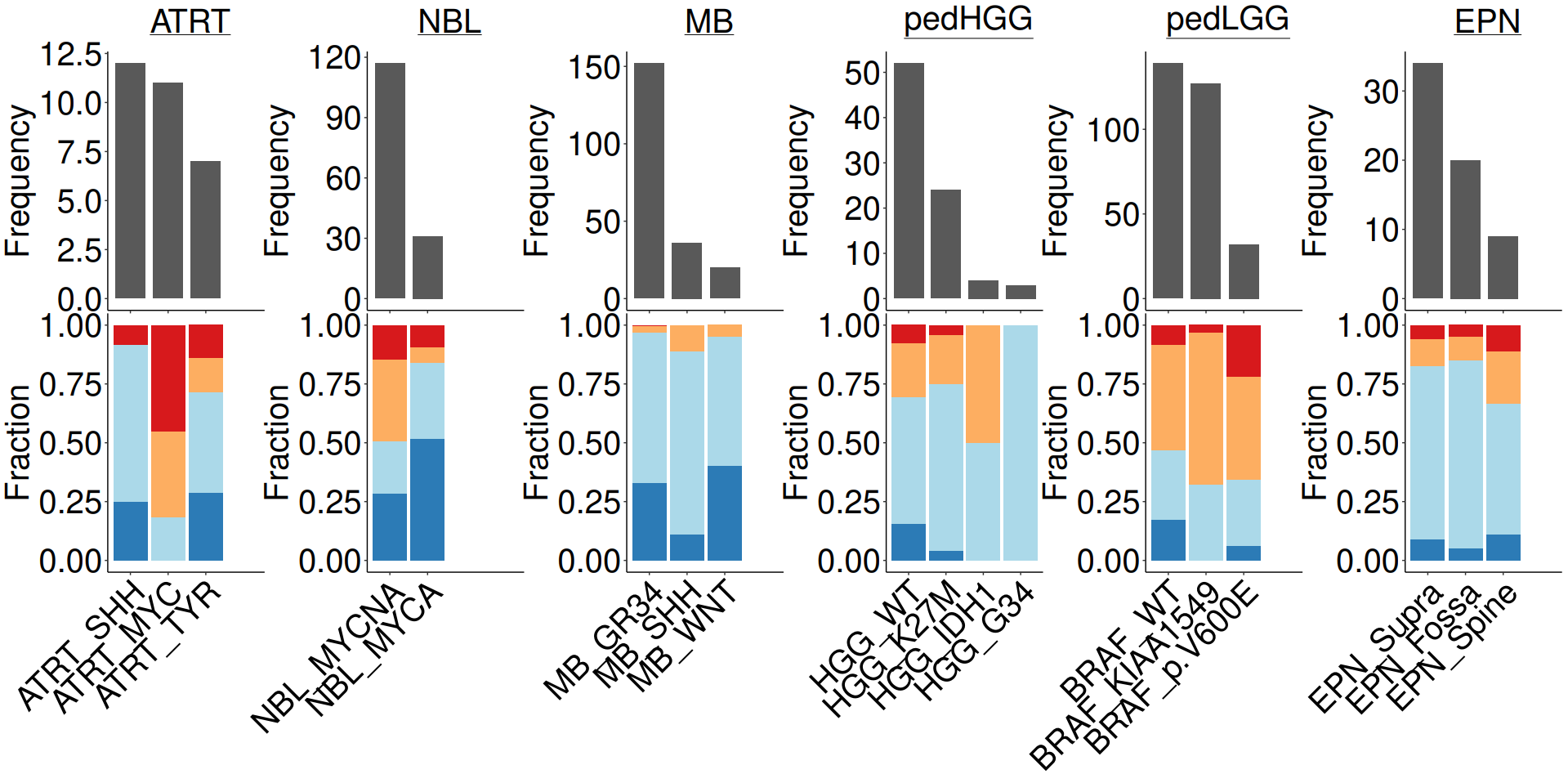
这是今天的最后一幅图啦,这幅图是一个显示不同类型癌症频率和分数的条形图。图中有六个类别的肿瘤,每个类别都有一个频率条形图和一个分数堆叠条形图,在条形图中,用灰色阴影表示每个类别的样本数,分数堆叠条形图则用多种颜色表示每个类别中不同亚型的百分比,每个亚型的缩写和代码在图的底部标注,例如,ATRT-TYR表示ATRT中的酪氨酸亚型,MB-WNT表示MB中的WNT信号通路亚型,这些亚型是根据基因组或转录组数据进行分析和分类的,反映了不同癌症类型的生物学特征和预后特征。
通过以上的分析,我们在pedNST中找到了四个广泛的免疫集群,并且进行了和免疫集群有关的相关性分析。
以上就是小果今天的分享啦,今天我们主要对儿童的一些肿瘤库数据进行了分析,相信大家一定对多数据库的分析有了新的思考!如果小伙伴们有任何疑问,欢迎关注小果,当然,也试一试我们的云生信小工具,只要输入合适的指令就可以直接绘制想要的图呢,链接:http://www.biocloudservice.com/home.html。