在进行单个细胞水平的分析时,单细胞转录组测序技术是非常便利高效的一种技术,它可以通过测量单个细胞中所有基因的表达水平,从而读取细胞的“日记”,这样我们就可以很容易的知道细胞在做什么,想什么,感受什么,以及其生命活动和什么有联系。
而在进行单细胞转录组测序分析的时候,我们常常绕不开给细胞进行注释这一步骤,也就是根据单细胞转录组数据,对不同的细胞聚类进行细胞类型的标识和命名,这一步中通常使用的是自动注释, 对自动注释而言,是一种利用算法和适当的先验生物学知识对细胞或细胞簇进行标记的方法,这种方法虽然高速有效,却存在错误注释的可能性。
基准研究表明,自动注释工具的性能是处于变化中的,它的精准度取决于被注释细胞类型的数据集和基因表达谱的独特性,比如说,使用自动注释区分T细胞和B细胞相对简单,但使用自动注释工具区分CD8+细胞毒性T细胞和自然杀伤细胞却比较困难。因此,我们需要对自动注释进行手动改进以及验证分析,从而让我们的注释结果更加准确可信。本次代码的数据量偏大,欢迎大家来租赁我们的服务器哦,关注公众号还可以获得独特的生信分析思路定制~
接下来小果将从头开始,带着小伙伴们一起完成单细胞注释的整个过程~本次教程我们使用的是细胞是:Peripheral blood mononuclear cells(PBMC)。这是外周血中具有单个核的细胞的混合群体,是免疫细胞的丰富来源,可以进一步纯化分离出多个细胞类型。
如图,是本次所需得基因文件。另外本次分析所需的R版本以及R包版本小果也帮大家整理好了~
Step 1 安装R包
install.packages(c(“BiocManager”,”Seurat”,”SCINA”,”ggplot2″,”devtools”,”shiny”))
BiocManager::install(c(“SingleCellExperiment”,”scater”,”dplyr”, “scmap”,
“celldex”,”SingleR”,”GSVA”,”S4Vectors”, “SummarizedExperiment”))
devtools::install_github(“immunogenomics/harmony”)
devtools::install_github(“romanhaa/cerebroApp”)
devtools::install_github(“mw201608/msigdb”)
Step 2 基于参考的自动注释
#从GEO数据库下载数据(由于已经下载好了所以直接导入文件即可)
set.seed(9742)
ref <- readRDS(“../data/data_DICE.RData”)
#格式化数据,确保与正在使用的工具兼容,这里使用的是scmap
# 在名为“cell_type1”的列中分配单元格类型标签
SummarizedExperiment::colData(ref)$cell_type1 <- SummarizedExperiment::colData(ref)$label.fine
# 在名为“feature_symbol”的列中分配基因名称
SummarizedExperiment::rowData(ref)$feature_symbol <- rownames(ref)
# 将数据转换为SingleCellExperiment对象
ref_sce <- SingleCellExperiment::SingleCellExperiment(assays=list(logcounts=Matrix::Matrix(SummarizedExperiment::assays(ref)$logcounts)),
colData=SummarizedExperiment::colData(ref),
rowData=SummarizedExperiment::rowData(ref))
这样参考数据就可以使用了,然后我们需要处理这些数据来构建将用来将未标记数据映射的索引。 首先,我们选择要使用的基因,这些基因将是在通过基因丢失分布将线性模型拟合到基因表达后被 scmap 检索的认为最具信息性的基因,信息最丰富的细胞具有高表达值和低丢失率。
#首先选择信息最丰富的特征来创建 scmap-cluster 参考
ref_sce <- scmap::selectFeatures(ref_sce, suppress_plot=FALSE)
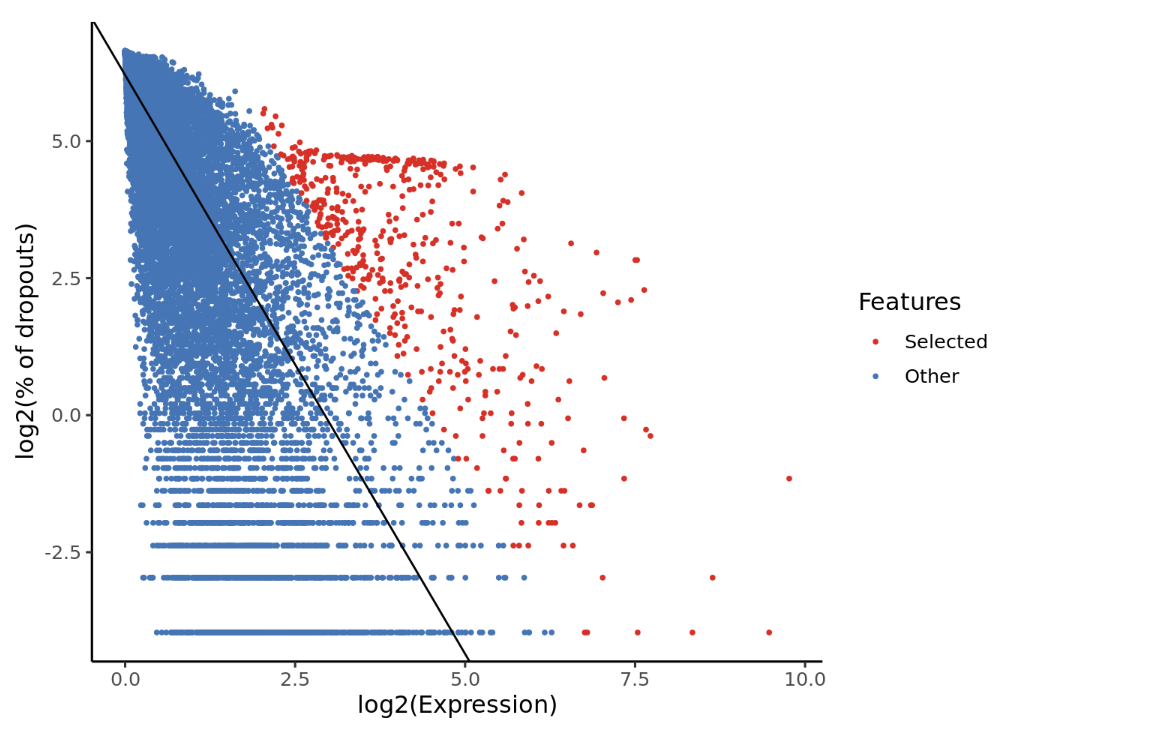
如图是通过可视化来展示单细胞转录组数据的质量控制,特征选择的结果。log2(Expression)表示每个特征的平均表达水平,log2(% of dropouts)表示每个特征在多少比例的细胞中没有被检测到。图中的红色点就是根据标准算法选择出来的特征,而蓝色点是被排除的特征。
#检查 scmap 选择的前 50 个基因
rownames(ref_sce)[which(SummarizedExperiment::rowData(ref_sce)$scmap_features)][1:50]
如图,是我们筛选的前  50个基因
50个基因
#通过检查基因名称向量的长度来检查选择了多少个基因
length(rownames(ref_sce)[which(SummarizedExperiment::rowData(ref_sce)$scmap_features)])

现在我们可以看到 scmap 选择使用的基因。 如果缺少关键标记基因,我们可以确保将它们包含在内,如下所示:
#创建要使用的关键标记的列
my_key_markers = c(“TRAC”, “TRBC1”, “TRBC2”, “TRDC”, “TRGC1”, “TRGC2”, “IGKC”)
# 确保标记位于 scmap 使用的功能列表中
SummarizedExperiment::rowData(ref_sce)$scmap_features[rownames(ref_sce) %in% my_key_markers] <- TRUE
# 通过再次检查基因名称向量的长度来检查是否添加了任何基因长度
length(rownames(ref_sce)[which(SummarizedExperiment::rowData(ref_sce)$scmap_features)])

我们可以删除可能是技术产物的基因,例如线粒体 RNA,如下所示:
# 从数据集中创建线粒体基因列表(以“MT”开头的基因)
mt_genes <- rownames(ref_sce)[grep(“^MT-“, rownames(ref_sce))]
# 从 scmap 使用的特征中删除这些基因
SummarizedExperiment::rowData(ref_sce)$scmap_features[rownames(ref_sce) %in% mt_genes] <- FALSE
# 检查有多少个基因
length(rownames(ref_sce)[which(SummarizedExperiment::rowData(ref_sce)$scmap_features)])

#提取特征并将其分配给新变量“scmap_feature_genes”
scmap_feature_genes <- rownames(ref_sce)[which(SummarizedExperiment::rowData(ref_sce)$scmap_features)]
#基因/特征的数量与我们刚刚检查的相同
length(scmap_feature_genes)

现在我们构建 scmap-cluster 中使用的参考配置文件,用于基于簇的细胞类型注释。我们 可以从 SingleCellExperiment 对象内部访问和绘制这些配置文件,如下所示:
# 创建参考资料;
# 生成参考配置文件后,scmap-cluster 不需要原始数据
ref_sce <- scmap::indexCluster(ref_sce)
# 将功能可视化为热图
# 重新格式化数据,以便它们可以用作 ggplot2 的输入
cormat <- reshape2::melt(as.matrix(S4Vectors::metadata(ref_sce)$scmap_cluster_index))
# 绘图
ggplot2::ggplot(cormat, ggplot2::aes(x = Var2, y = Var1, fill = value)) +
ggplot2::geom_tile() +
ggplot2::scale_fill_gradient2(low = “blue”, high = “darkred”,
name = “Expression value”) +
ggplot2::theme_minimal() +
ggplot2::theme(axis.text.x = ggplot2::element_text(angle = 90, vjust = 1,
size = 18, hjust = 1),
axis.text.y = ggplot2::element_text(size = 15),
axis.title.x = ggplot2::element_blank(),
axis.title.y = ggplot2::element_blank())
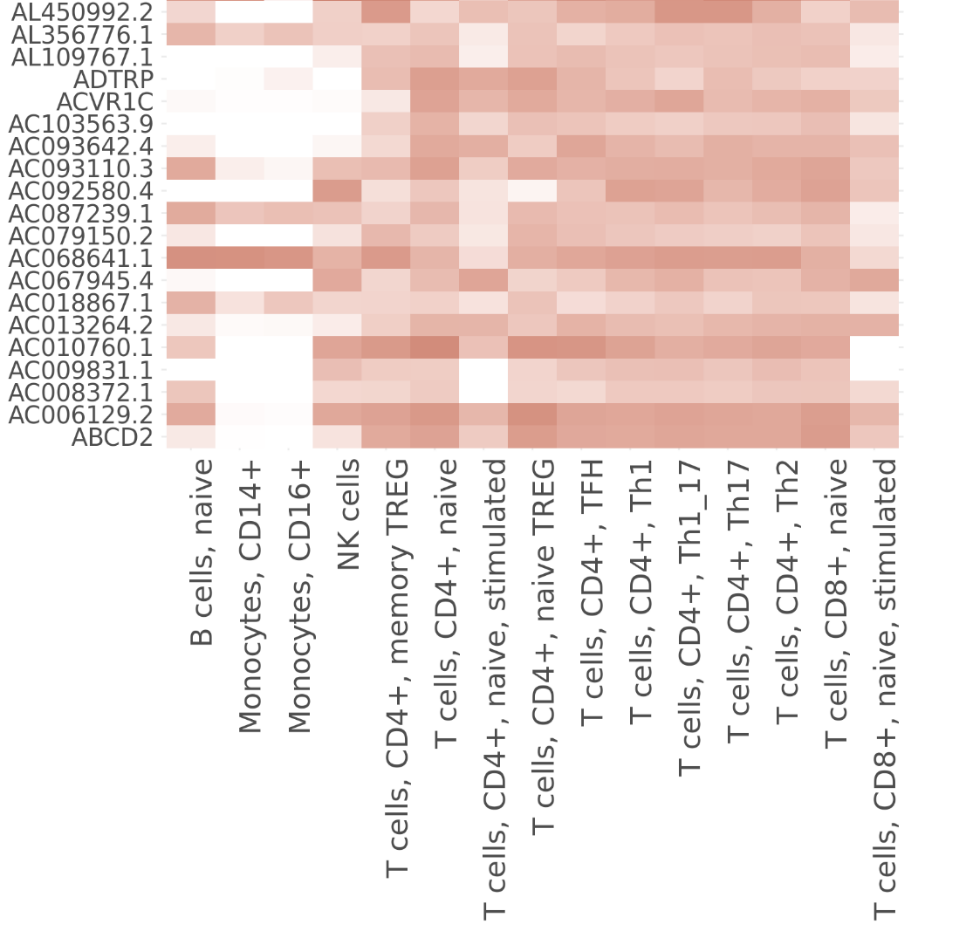
结果如图所示,这个图片显示了不同类型的细胞与特定基因之间的关系。颜色的深浅表示关联程度或活动水平的高低。小果这里只截取了一部分,完整的图片大家可以自己试着绘制哦~
接下来我们用scmap-cell 来注释数据集中的单个细胞。
# 将表达式信息存储为变量
scmap_cluster_reference <- S4Vectors::metadata(ref_sce)$scmap_cluster_index
# 更新之前的引用以也包含 scmap-cell 引用
ref_sce <- scmap::indexCell(ref_sce)
#从引用中提取 scmap 索引并将其存储为变量
scmap_cell_reference <- S4Vectors::metadata(ref_sce)$scmap_cell_index
# 从引用中提取关联的细胞 ID 并保存为变量
scmap_cell_metadata <- SummarizedExperiment::colData(ref_sce)
然后将查询数据集中的细胞分配给参考数据集,利用一个已有的参考数据集来对一个新的查询数据集进行注释或映射。我们将使用的查询数据集由 10X genomics 提供,大家可以从小果给大家提供的链接下载~
data <- Seurat::Read10X(“../data/hg19/”)
# 从原始矩阵中进行 SingleCellExperiment
query_sce <- SingleCellExperiment::SingleCellExperiment(assays=list(counts=data))
# 从 Seurat 对象制作 SingleCellExperiment
query_seur <- Seurat::CreateSeuratObject(data)
query_sce <- Seurat::as.SingleCellExperiment(query_seur)
# 使用 scatter 包标准化数据
query_sce <- scater::logNormCounts(query_sce)
# 添加feature_symbol列(即基因符号)
SummarizedExperiment::rowData(query_sce)$feature_symbol <- rownames(query_sce)
现在,我们已准备好使用 scmap-cluster 对数据进行注释。
# 运行 scmapCluster
scmap_cluster_res <- scmap::scmapCluster(projection=query_sce,
index_list=list(immune1 = scmap_cluster_reference),
threshold=0.1)
# 绘制注释结果
par(mar=c(13, 4, 1, 0))
barplot(table(scmap_cluster_res$combined_labs), las=2)
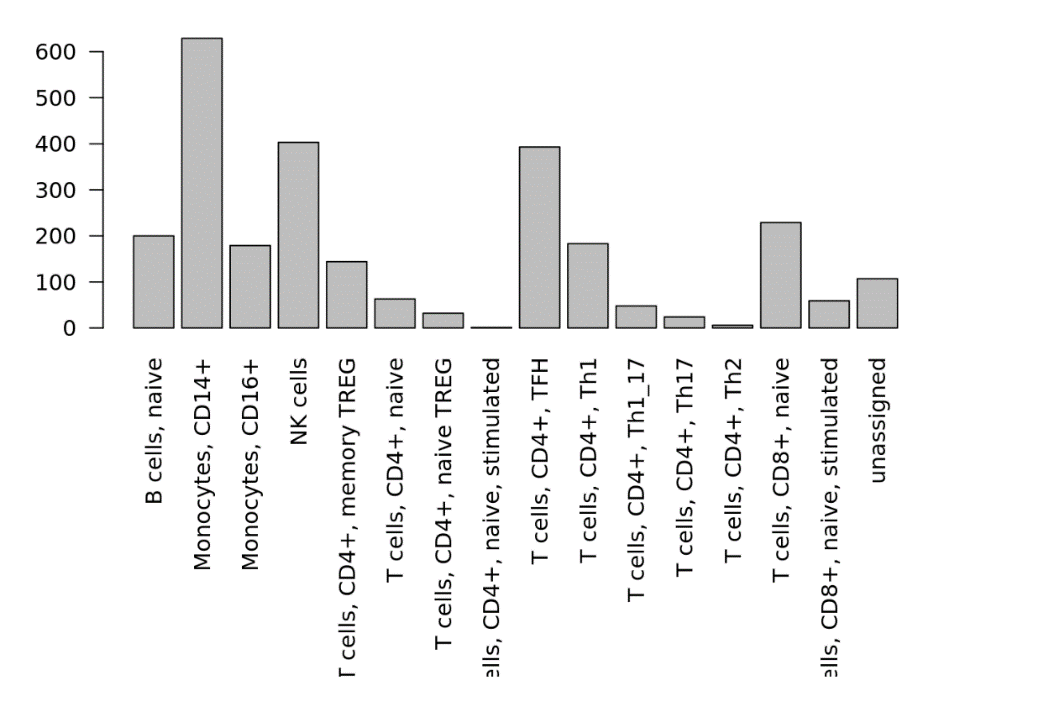
上图就是我们进行注释的结果啦,可以看到Monocytes CD14+细胞的数量是最多的。
# 将此注释信息存储在查询对象中
SummarizedExperiment::colData(query_sce)$scmap_cluster <- scmap_cluster_res$combined_labs
# 制作单元格的 UMAP,并使用 scmapCluster 中的单元格类型注释进行标记
query_sce <- scater::runUMAP(query_sce)
scater::plotReducedDim(query_sce, dimred=”UMAP”, colour_by=”scmap_cluster”)
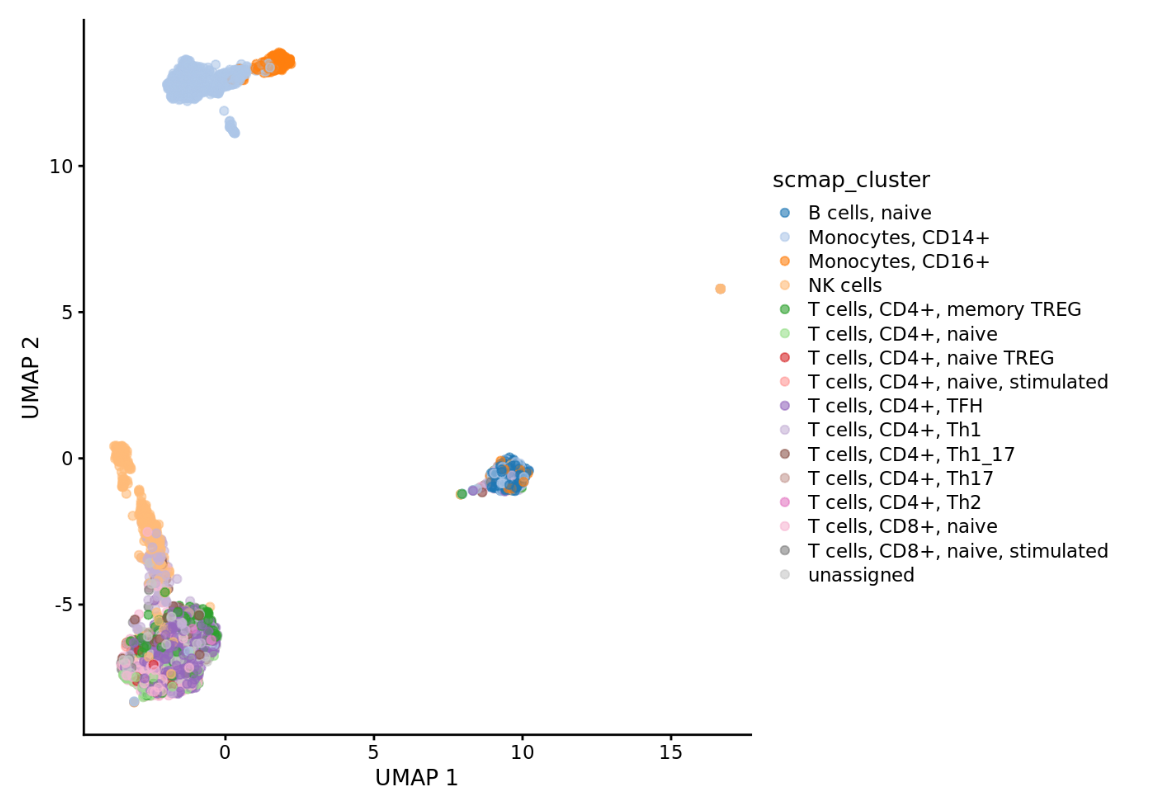
结果如图,展示了不同类型的细胞在二维空间中的分布和聚类。每种颜色代表一种特定类型的细胞,例如B细胞、T细胞等。
除此之外,我们还可以用scmap_cell,singleR,Harmony_lab等方式进行自动注释~
Step 3 细化注释
在这里我们使用跨工具的最常见标签来分配最终的自动注释标签。
annotation_columns <- c(“scmap_cluster”, “scmap_cell”, “singleR”, “Harmony_lab”)
get_consensus_label <- function(labels){
labels <- labels[labels != “ambiguous”]
if (length(labels) == 0) {return(“ambiguous”)}
freq <- table(labels)
label <- names(freq)[which(freq == max(freq))]
if (length(label) > 1) {return(“ambiguous”)}
return(label)
}
SummarizedExperiment::colData(query_sce)$consensus_lab <- apply(SummarizedExperiment::colData(query_sce)[,annotation_columns], 1, get_consensus_label)
scater::plotReducedDim(query_sce, dimred=”UMAP”, colour_by=”consensus_lab”)
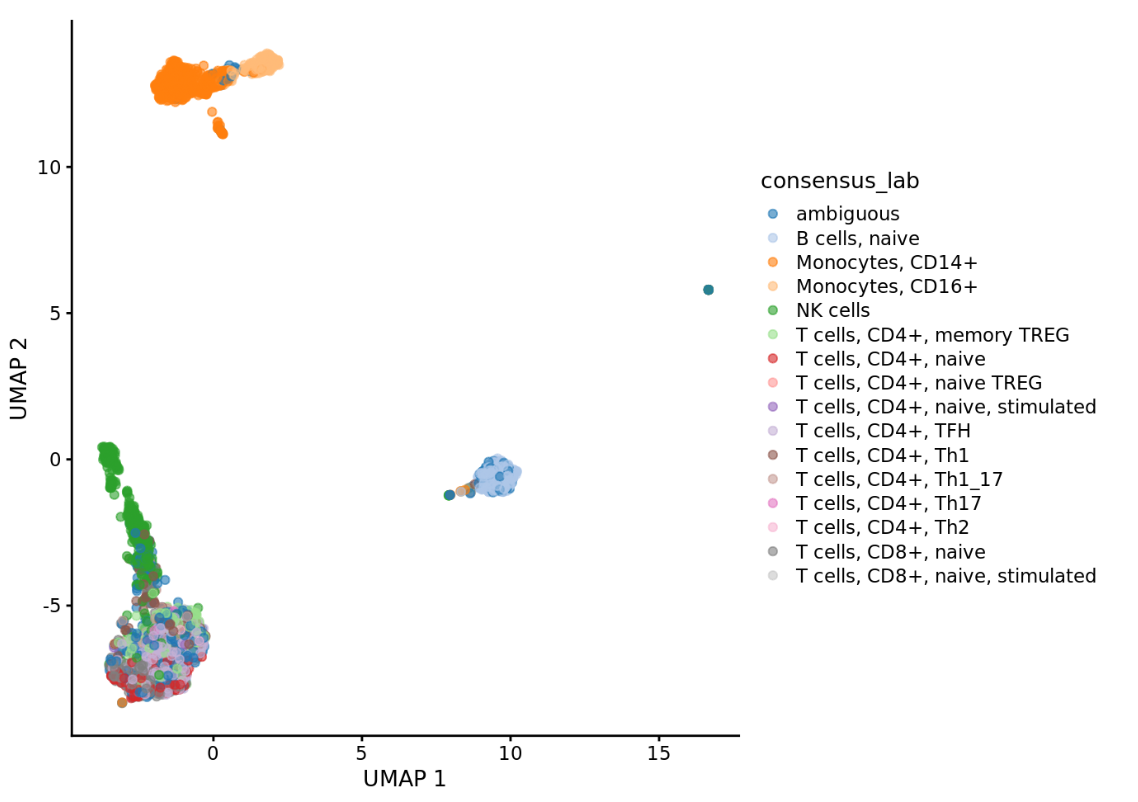
细化的结果如图,可以从侧边的标注看出来明显比之前的分类更加细致了。
Step 4 手动注释
首先我们需要检索标记基因,如果没有每种细胞类型的标记物的列表,也没有高质量的参考数据集的话,可以从查询数据的每个聚类中提取顶级标记基因。这里我们使用 Seurat来完成,将数据格式化为Seurat 对象。先对数据进行归一化和缩放,并且确定细胞之间的可变基因。
# 使用 Seurat 的 SCTransform 函数缩放和标准化数据
query_seur <- Seurat::SCTransform(query_seur)
#接下来,对数据执行不同类型的降维,以便可以在二维空间中将单元格组合在一起。
query_seur <- Seurat::RunPCA(query_seur)
query_seur <- Seurat::RunTSNE(query_seur)
query_seur <- Seurat::RunUMAP(query_seur, dims = 1:50)
#以选定的分辨率对数据进行聚类
# 确定每个单元格的“最近邻居”
query_seur <- Seurat::FindNeighbors(query_seur, dims = 1:50)
# 聚类
query_seur <- Seurat::FindClusters(query_seur, resolution = 0.5)
#可视化
Seurat::DimPlot(query_seur, reduction = “UMAP”)
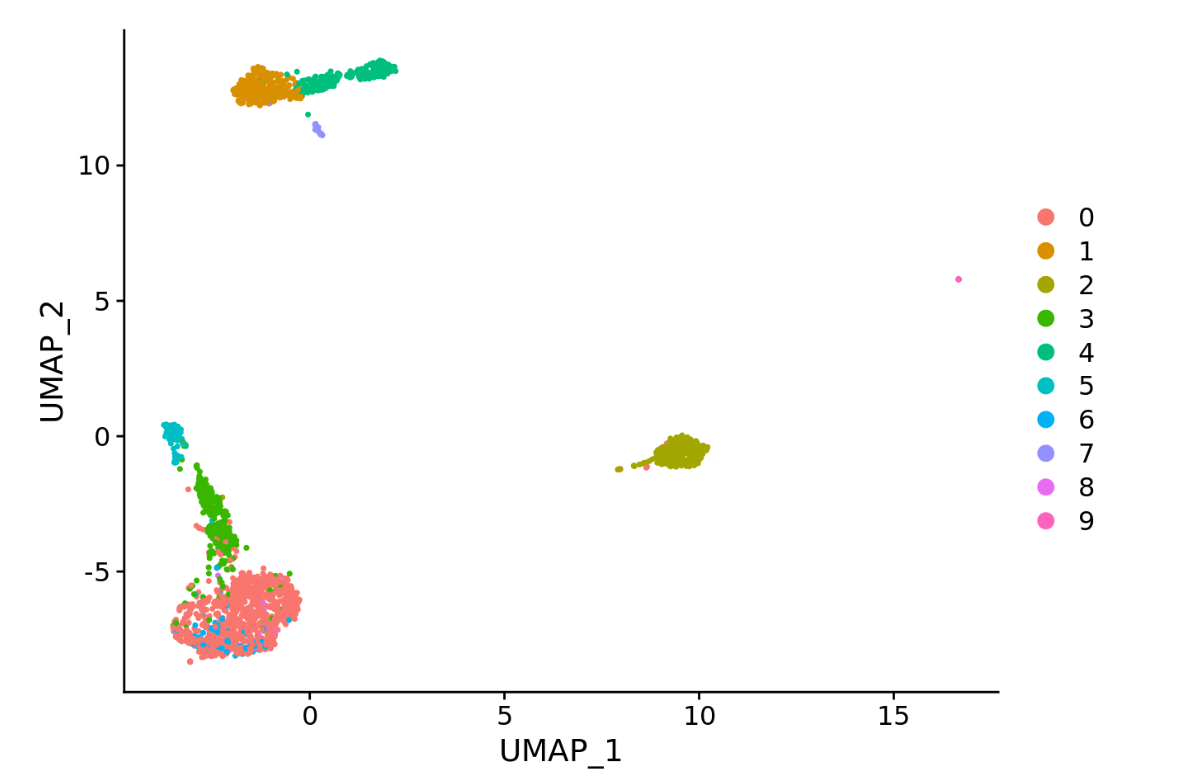
如图,是还没有提取标记基因的初步绘制的热图,接下来我们提取顶部基因。
#使用 Seurat 中的 FindAllMarkers 函数来完成
markers_seur <- Seurat::FindAllMarkers(query_seur, only.pos = TRUE)
# 查看标记,它们按照每个簇的 p 值从最低到最高的顺序排列
markers_seur
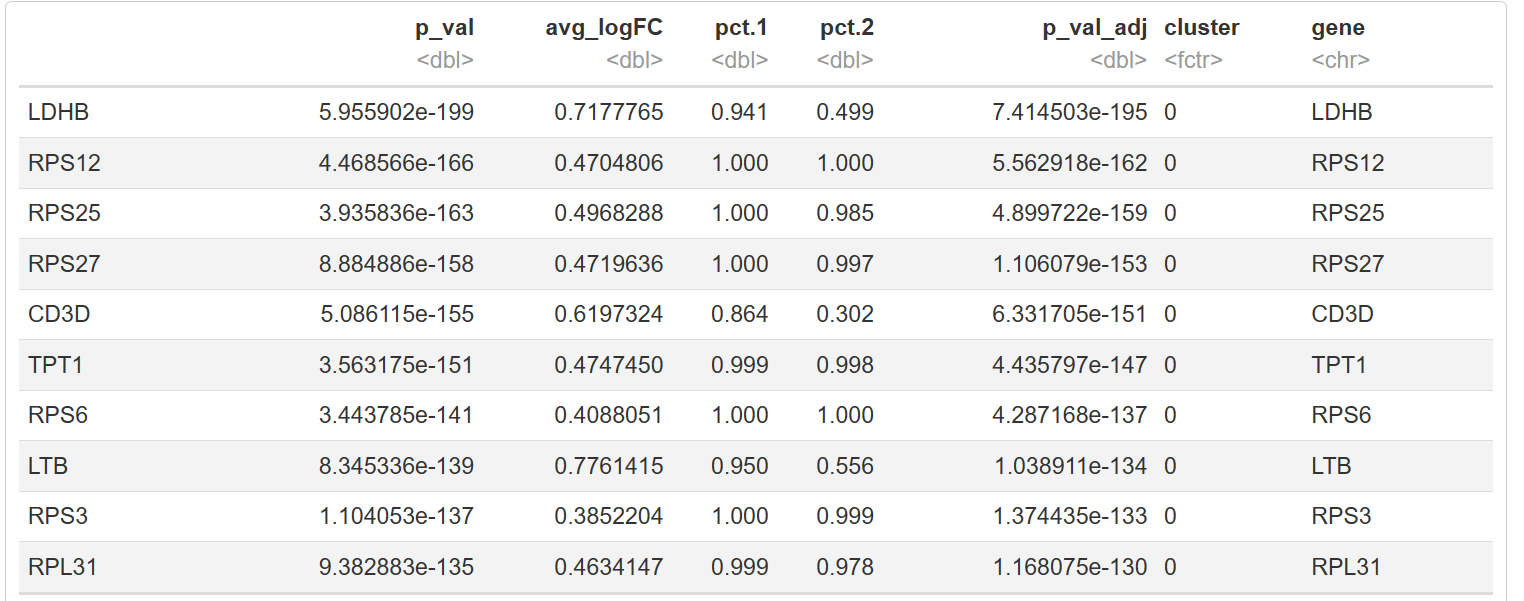
结果如图,是我们挑选的标记基因。不同细胞簇之间的标记基因的表达通常用点图或热图来展示。点图显示了每个细胞簇中表达某个标记基因的细胞的百分比(点的大小)和该细胞簇的平均检测到的基因表达水平。热图显示了不同细胞簇的平均标记基因表达水平。这两种图都是用Seurat创建的。
require(dplyr)
# 检索每个簇的前 5 个标记基因
# 使用 AVG_LOG 列下具有最高值的基因
top5 <- markers_seur %>% group_by(cluster) %>%
dplyr::slice_max(get(grep(“^avg_log”, colnames(markers_seur), value = TRUE)),
n = 5)
# 绘制点图
Seurat::DotPlot(query_seur, features = unique(top5$gene)) +
ggplot2::theme(axis.text.x = ggplot2::element_text(angle = 90, vjust = 1,
size = 8, hjust = 1)) +
Seurat::NoLegend()
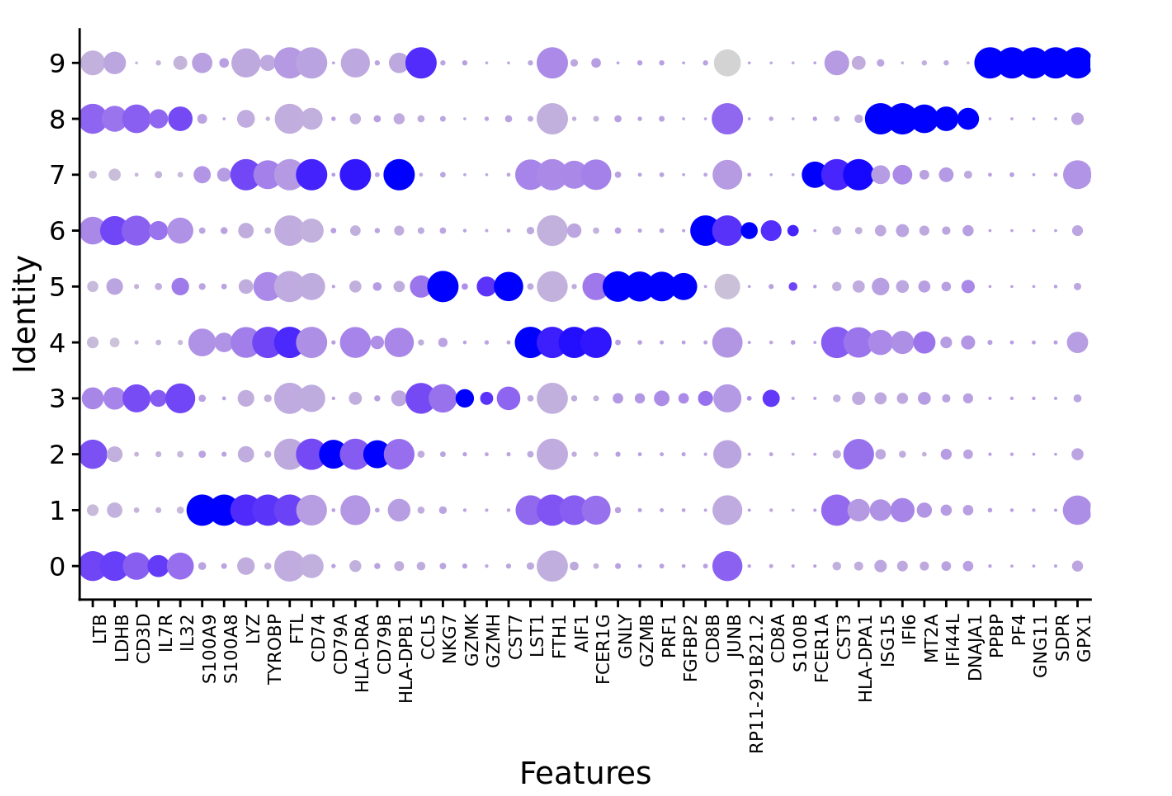
如图所示,清晰的显示了每个细胞簇中表达某个标记基因的细胞的百分比(点的大小)和该细胞簇的平均检测到的基因表达水平。
# 绘制热图
Seurat::DoHeatmap(query_seur, features = unique(top5$gene)) +
Seurat::NoLegend() +
ggplot2::theme(axis.text.y = ggplot2::element_text(size = 8))
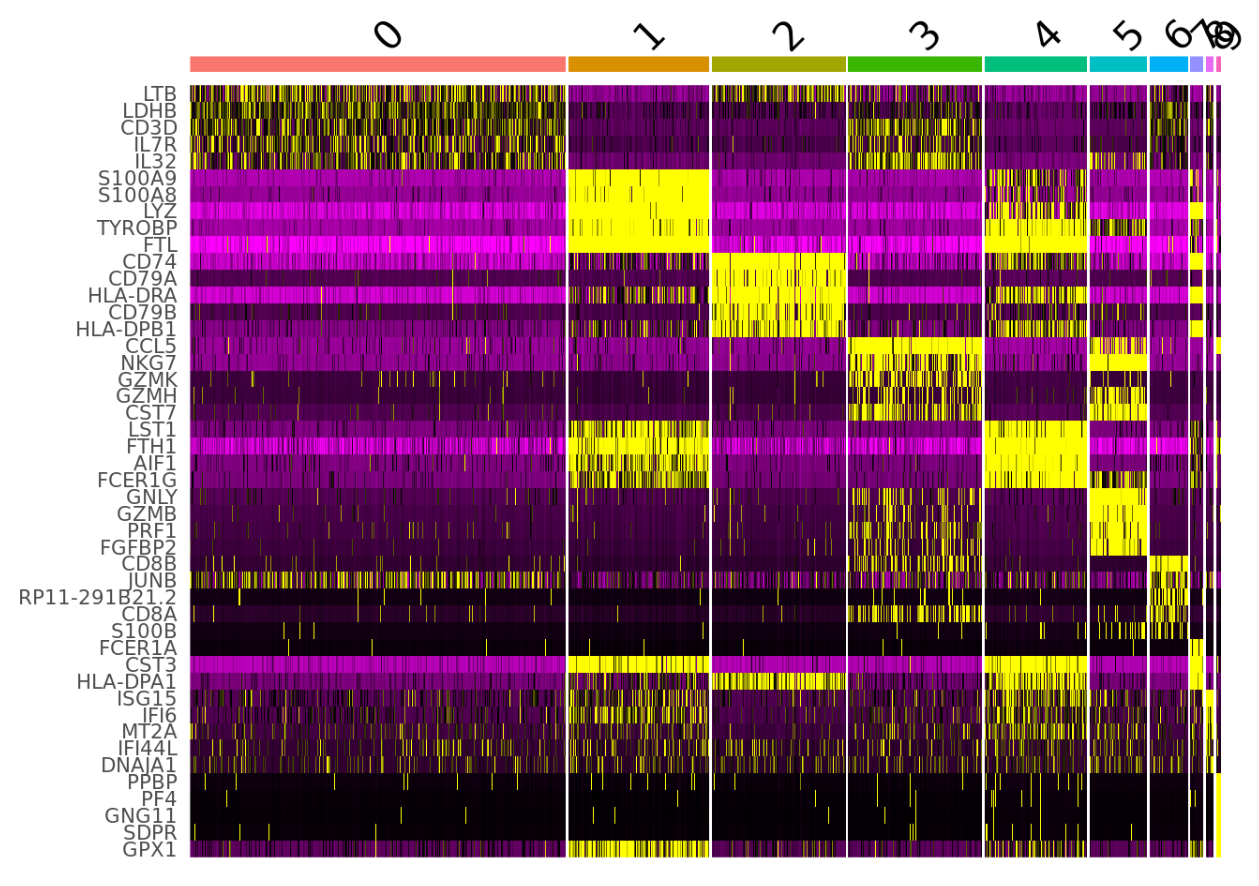
如图所示,显示了不同细胞簇的平均标记基因表达水平。
通过上述的工具和方法可以为scRNA-seq数据提供可靠的细胞类型标记。然而,由于算法的浮动以及标记的模糊性,使用其他的方法(例如统计方法)或进行实验来确认单元格注释标签是非常重要的。此外,由于mRNA测量只能部分定义细胞类型和功能,因此关于新细胞类型的重要结论必须经过实验验证。
例如,可以使用T细胞受体(TCR)和B细胞受体克隆分型来改进组织驻留免疫细胞的细胞类型标记,以检查T细胞和B细胞的转录特征,分别由它们表达的TCR和B细胞受体分层。
以上就是小果今天给大家带来的全部内容啦!单细胞注释是单细胞的转录组分析中非常重要的一步,无论是自动注释还是手动选择标记基因进行注释,都具有一定的不确定性,但是我们可以通过将多种自动注释以及手动注释的结果进行整合,使得自己得分析数据做到相对的精准。如果小伙伴们有任何疑问,欢迎关注小果,当然,也可以试一试我们的云生信小工具,只要输入合适的指令就可以直接绘制想要的图呢,链接:http://www.biocloudservice.com/home.html。