各位小伙伴大家好啊,又到了小果的代练时间啦,是不是很激动呢?让我们这就开始今天的问题吧~
昨晚小果在整理自己的工作内容的时候,看到同事在对已获得部分研究结果的基因组数据进行分析,问题情境是:要具现多个染色体上,个别基因的位置关系,并根据其基因长度和位置间隙等比例缩放。然后同事就使用计算器进行等比例缩放,使用AI进行21个染色体,一共143个基因的绘制。幸亏最后小果及时出手,要不然同事这个班加的太“亏”了。所以今天,小果想把这个R包分享给各位小伙伴,以便减轻小伙伴们的工作压力和美化我们的结果。
今天这个能给大家带来方便的R包叫做gggenes,没错!看名字就知道它是ggplot2包的拓展包,因此它能完美继承各种ggplot2包的基础函数,达到高度的调控性,因此能可视化出效果较好的基因结构图,这种图形通常显示基因的外显子和内含子,以及各转录本的基因结构信息。
所以,要进行该基因结构图的绘制,不能避免的要对基因结构进行研究,而对于这种针对测序数据结构文件的处理,小果推荐使用服务器进行,因为有多个软件是针对Linux系统进行编写,对大数据的处理更加友好,如果各位小伙伴现在没有服务器的话,可以联系小果进行租赁哦~
那下面就开始我们今天的代练吧!
#BiocManager::install(“wilkox/gggenes”)
library(gggenes)
library(ggplot2)
library(RColorBrewer)
的确有的包可以通过install.packages(‘’)命令进行安装,但这是在CRAN中原本就已经存在的,gggenes为个人编写,所以可以在Github上进行下载安装,其中‘/’前的‘wilkox’为编写人的仓库名称,后面则为包的名称。
display.brewer.all() #是RcolorBrewer的函数,能查看该包中所有的色块,对画图时颜色的选取有帮助
#数据读入
data <- read.table(‘gggenes.txt’,header = T,sep = ‘\t’)
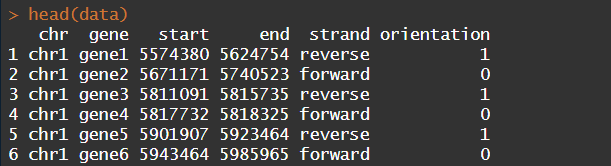
head(data)
chr代表分别是哪几条染色体,gene代表的是某一染色体上的哪个基因,star和end表示基因的起始与终止并用strand表示基因的方向。
subdata <- read.table(‘subgene.txt’,header = T,sep=’\t’)
head(subdata)
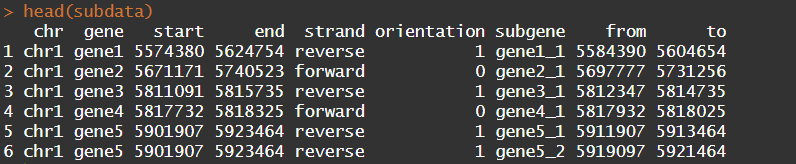
大家可以看到,subdata相较于data而言多了‘subgene’,‘from’和‘to’这三列,分表表示,某染色体上某基因的转录本或内含子的起始和终止位点。
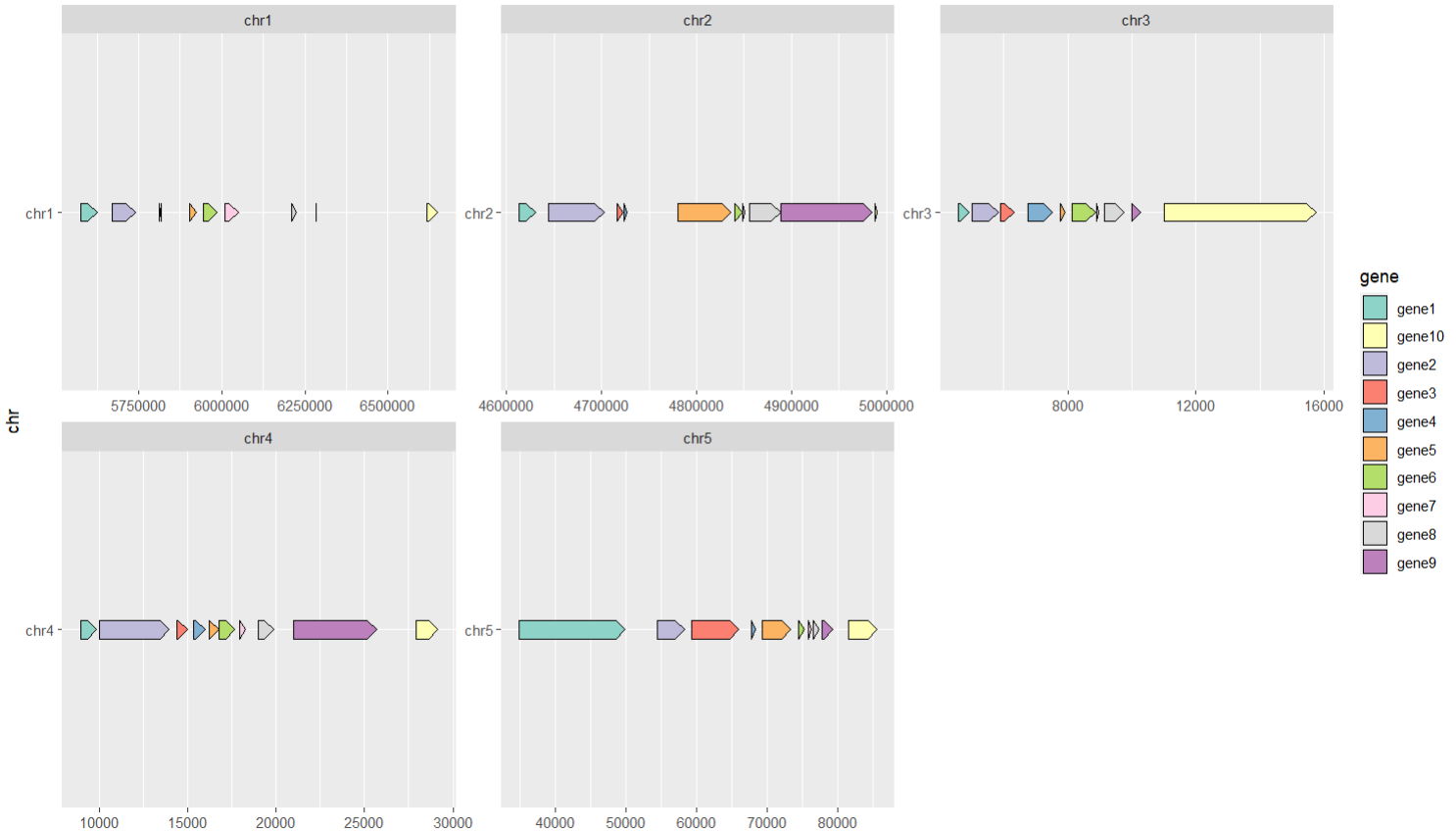
#简单出图
P1 <- ggplot(data , aes(xmin = start, xmax = end, y = chr, fill = gene)) +
geom_gene_arrow(arrowhead_width = grid::unit(2, “mm”), #对箭头的头的宽度进行调整
arrowhead_height = grid::unit(4, “mm”), #对箭头的头的高度进行调整
arrow_body_height = grid::unit(4, “mm”),) + #对箭头的体宽进行调整
facet_wrap(~ chr, #可以是变可以是公式,代表根据chr这一变量分别绘制一张图
scales = “free”, #有fixed和free两种,控制的是每张图的刻度数,free表示每个图有独立的刻度
ncol = ) + #在屏幕上的展示方式有ncol和nrow两个函数控制
scale_fill_brewer(palette = “Set3”)
P1
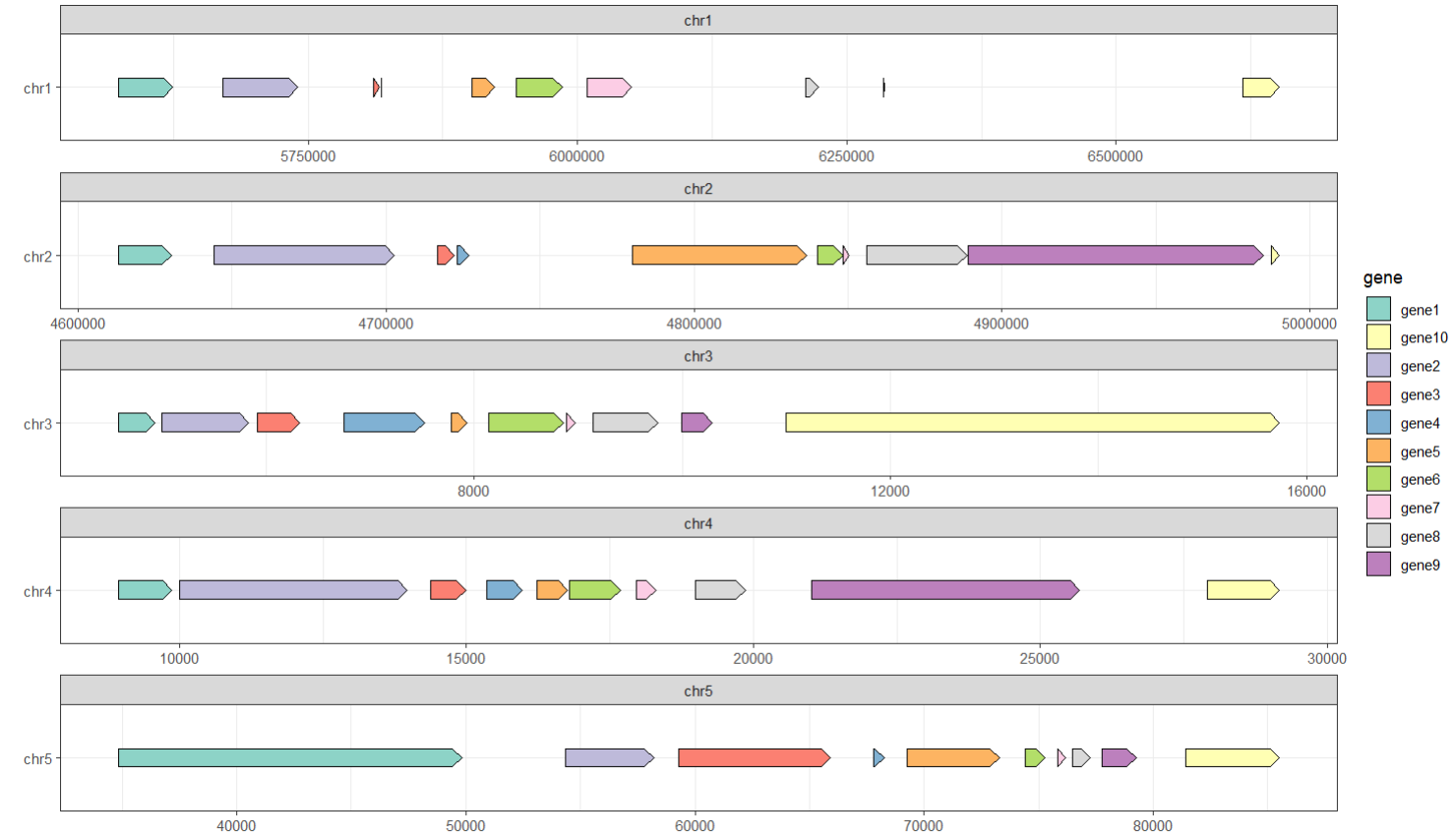
#更改主题
P2 <- ggplot(data , aes(xmin = start, xmax = end, y = chr, fill = gene)) +
geom_gene_arrow(arrowhead_width = grid::unit(2, “mm”), #对箭头的头的宽度进行调整
arrowhead_height = grid::unit(4, “mm”), #对箭头的头的高度进行调整
arrow_body_height = grid::unit(4, “mm”),) + #对箭头的体宽进行调整
facet_wrap(~ chr, #可以是变可以是公式,代表根据chr这一变量分别绘制一张图
scales = “free”, #有fixed和free两种,控制的是每张图的刻度数,free表示每个图有独立的刻度
ncol = 1) + #在屏幕上的展示方式有ncol和nrow两个函数控制
scale_fill_brewer(palette = “Set3″) +
ylab(”) +
theme_bw() #因为该函数是ggplot2的拓展,所以可以通过theme函数自定义,theme_gene是其自带函数
P2
#添加gene标签
P3 <- ggplot(data , aes(xmin = start, xmax = end, y = chr, fill = gene, label = gene)) +
geom_gene_arrow(arrowhead_width = grid::unit(2, “mm”), #对箭头的头的宽度进行调整
arrowhead_height = grid::unit(4, “mm”), #对箭头的头的高度进行调整
arrow_body_height = grid::unit(4, “mm”),) + #对箭头的体宽进行调整
facet_wrap(~ chr, #可以是变可以是公式,代表根据chr这一变量分别绘制一张图
scales = “free”, #有fixed和free两种,控制的是每张图的刻度数,free表示每个图有独立的刻度
ncol = 1) + #在屏幕上的展示方式有ncol和nrow两个函数控制
scale_fill_brewer(palette = “Set3”) +
geom_gene_label(align = ‘left’) + #标签角度’centre’,’left’ and ‘right’.
theme_genes() #因为该函数是ggplot2的拓展,所以可以通过theme函数自定义,theme_gene是其自带函数
P3
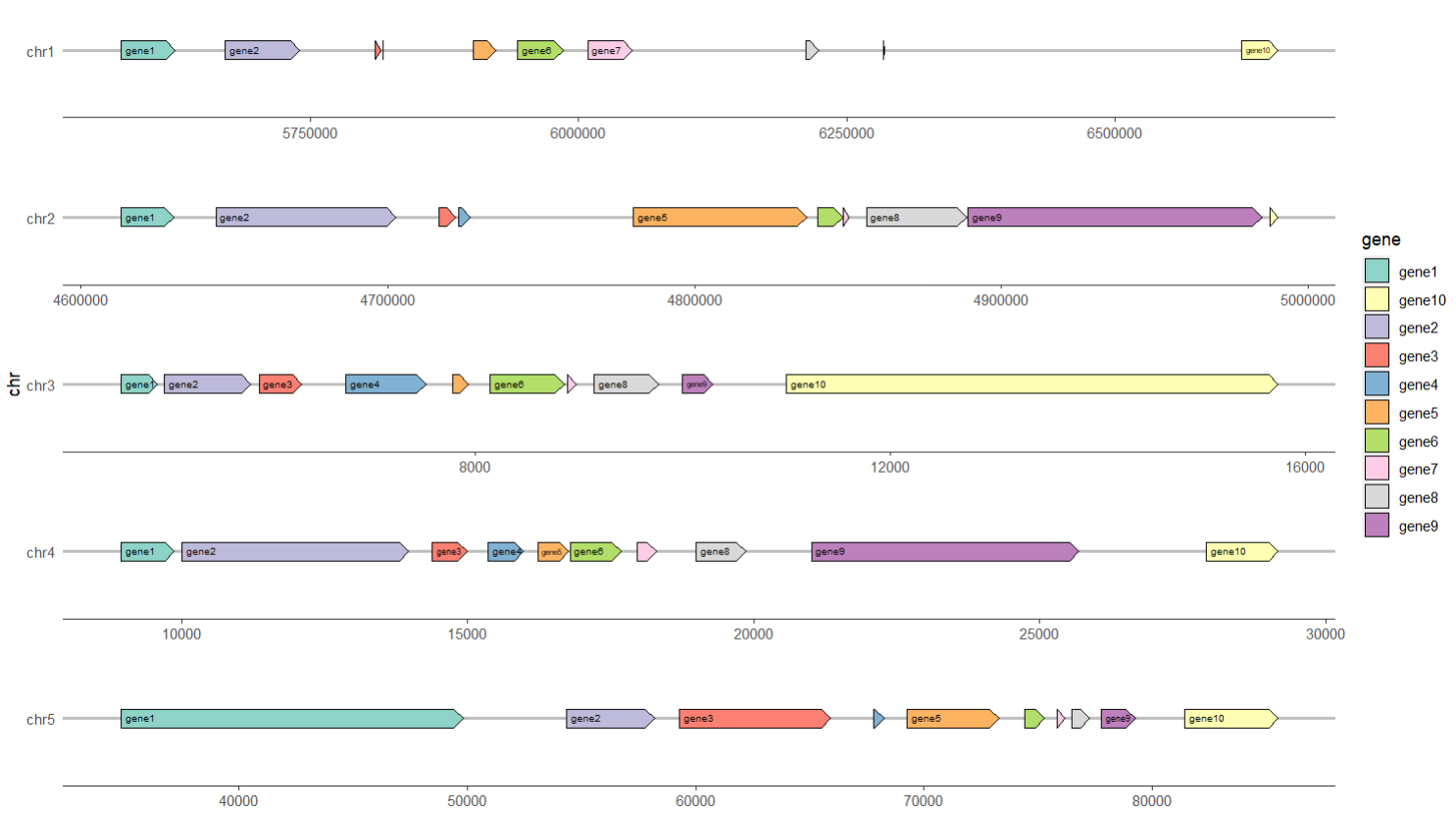
#按照基因方向进行翻转
P4 <- ggplot(data , aes(xmin = start, xmax = end, y = chr, fill = gene, label = gene,forward = orientation)) +
geom_gene_arrow(arrowhead_width = grid::unit(4, “mm”), #对箭头的头的宽度进行调整
arrowhead_height = grid::unit(4, “mm”), #对箭头的头的高度进行调整
arrow_body_height = grid::unit(4, “mm”),) + #对箭头的体宽进行调整
facet_wrap(~ chr, #可以是变可以是公式,代表根据chr这一变量分别绘制一张图
scales = “free”, #有fixed和free两种,控制的是每张图的刻度数,free表示每个图有独立的刻度
ncol = 1) + #在屏幕上的展示方式有ncol和nrow两个函数控制
scale_fill_brewer(palette = “Set3”) +
geom_gene_label(align = ‘centre’) +
theme_genes() #因为该函数是ggplot2的拓展,所以可以通过theme函数自定义,theme_gene是其自带函数
P4
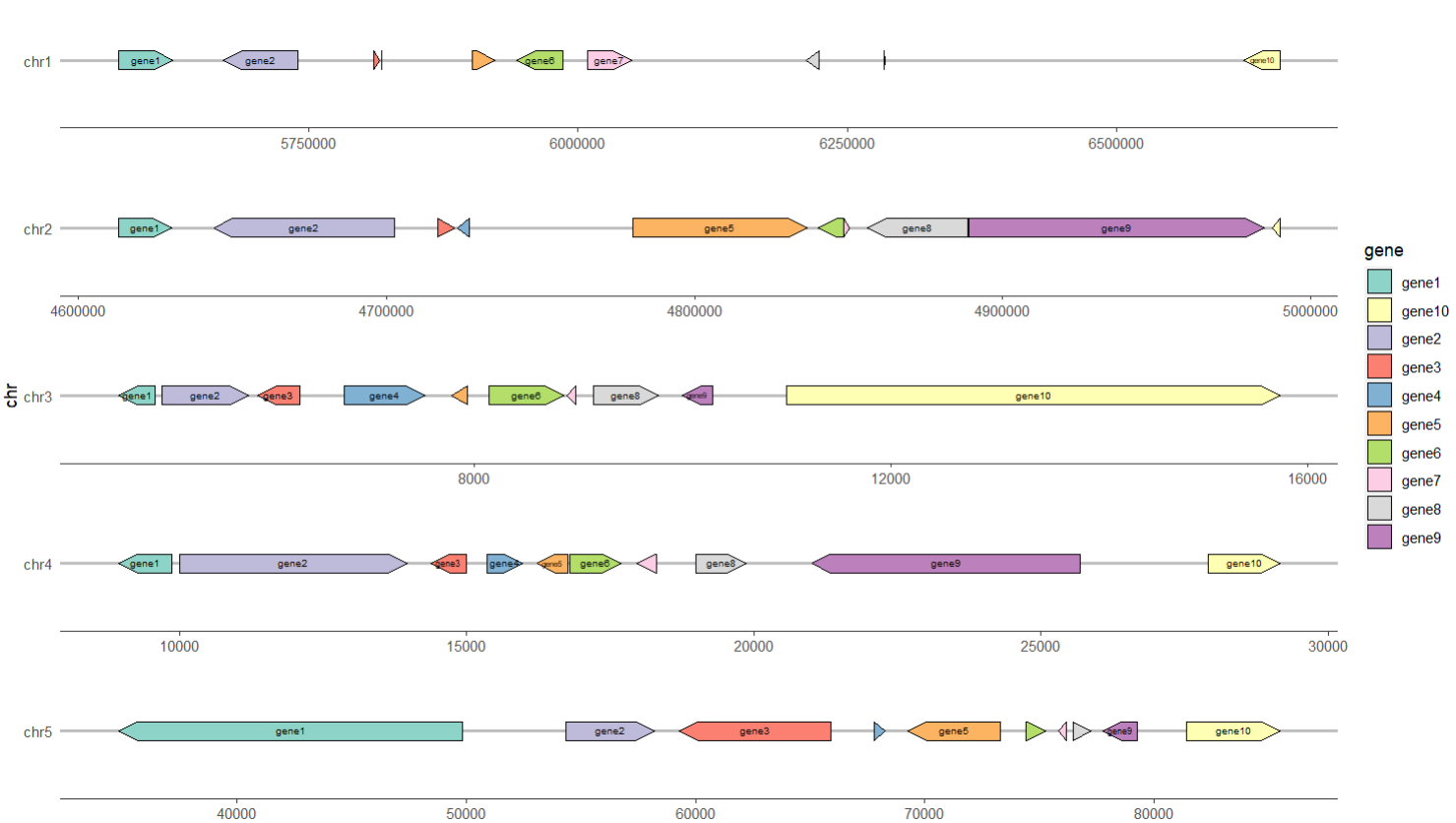
#对个别基因的内部结构做标识
P5 <- ggplot(subdata , aes(xmin = start, xmax = end, y = chr, forward = orientation)) +
geom_gene_arrow(arrowhead_width = grid::unit(4, “mm”), #对箭头的头的宽度进行调整
arrowhead_height = grid::unit(4, “mm”), #对箭头的头的高度进行调整
arrow_body_height = grid::unit(4, “mm”),) + #对箭头的体宽进行调整
geom_subgene_arrow(data = subdata,aes(xmin = start, xmax = end,y = chr,fill = subgene,
xsubmin = from,xsubmax = to),color = ‘black’) +
facet_wrap(~ chr, #可以是变可以是公式,代表根据chr这一变量分别绘制一张图
scales = “free”, #有fixed和free两种,控制的是每张图的刻度数,free表示每个图有独立的刻度
ncol = 1) + #在屏幕上的展示方式有ncol和nrow两个函数控制
scale_fill_brewer(palette = “Set3″) +
theme_bw() +
ylab(”)
P5
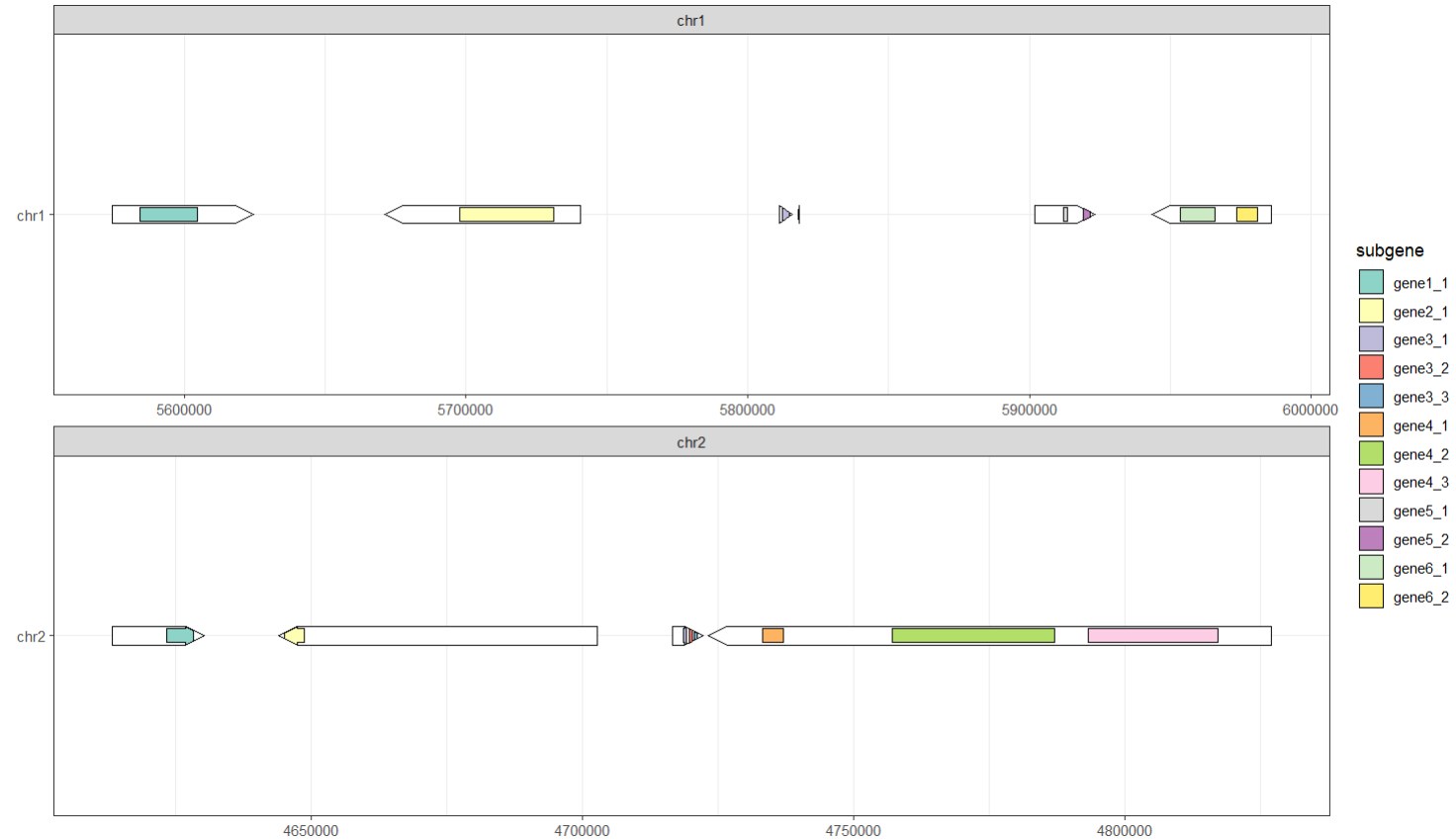
好啦,以上就是今天全部的代码代练哦,是不是很简单呢,其中前4张图用到的数据是data,最后一张图用到的数据是subdata。不知道大家有没有注意,在最后绘图的时候ggplot后面的aes中被小果删除了fill这个选项,而在后面的geom_subgene_arrow的函数中添加了fill = subgene,这是因为如果不删除ggplot中的填充映射的话会导致整个基因都被填充上颜色,而导致无法分辨某个基因的内部结构。
其实,基因结构图可适用于多个场合,包括基因注释、结构变异和质粒构建等,说明这还是很重要的。但如果各位小伙伴并不想在图形可视化上下太大的功夫,可以来小果的云生信平台,里面有多个已经成熟的代码,能帮助各位小伙伴火速出图,且支持字体和颜色的定制哦!快来加入我们吧。
http://www.biocloudservice.com/home.html