Hello,各位小伙伴大家好哦,很高兴又到分享知识的时间啦!
众所周知,转录组是生物学领域中一种研究基因表达的方法,是指基因中的信息被转录成RNA,并在随后翻译成蛋白质的过程。转录组学能帮助大家理解细胞或组织中所有基因的总体转录情况,从而在揭示基因的调控、发现新的RNA分子以及理解基因在不同生理和病理条件下的表达变化有所帮助。
针对有参转录组,大家的确可以使用clusterProfiler包进行GO富集分析,只需要获得你想富集分析的基因名称就OK了。但如果是无参转录组呢,该怎么办呢?咱们没有已经确定的gene symbol,只是拥有公司给出的组装基因的名称,这并不能在数据库中被识别,所以我们只能通过本地建库的方法,来进行无参转录组的富集分析了。
我们本地建库的配置文件有两个,一个是包含基因ID和基因注释的gene_info文件,一个是包含基因ID和GO号及EVIDENCE行的gene2go文件。
咱们今天所用数据较大,小果推荐大家使用服务器进行分析,如果各位小伙伴没有服务器的话,可以联系小果进行租赁哦~
现在咱们就进行两个配置文件的制作吧!另外提一句,今天使用的数据是经过eggNOG注释后的数据,大家可以参照最后的输出结果,对自己的无参转录组数据进行处理哦~
#调用包
library(clusterProfiler)
library(pheatmap)
library(readr)
library(tidyverse)
library(org.Hs.eg.db)
library(pathview)
#差异富集分析总数
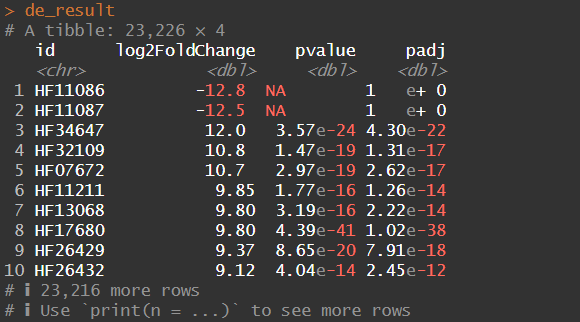
de_result <- read_delim(‘结果文件.tsv’)
de_result
#eggnog富集分析结果筛选
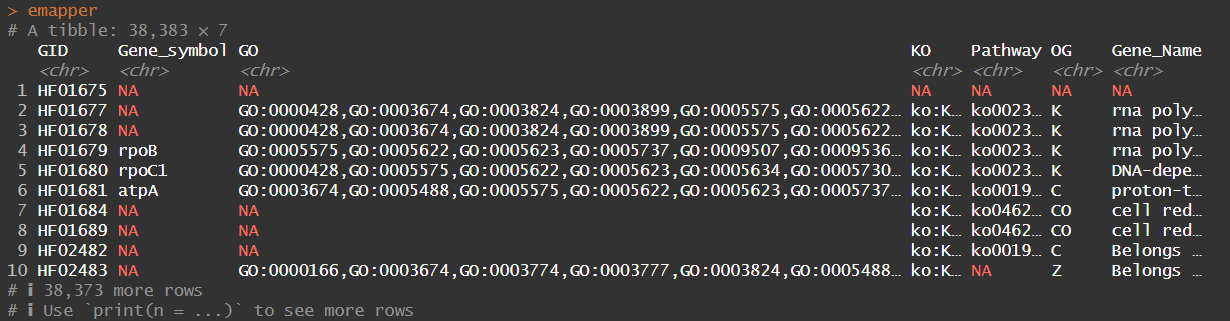
emapper <- read_delim(‘query_seqs.fa.emapper.annotations’,
‘\t’,escape_double = FALSE,col_names = FALSE,
comment = “#”,trim_ws = TRUE) %>%
dplyr::select(GID = X1,
Gene_symbol = X6,
GO = X7,
KO = X9,
Pathway = X10,
OG = X21,
Gene_Name = X22)
emapper
#筛选要富集基因
gene <- filter(de_result,abs(log2FoldChange) > 2 & padj < 0.01) %>%
pull(id)
#将所有基因的log2FC以基因名为名称转化成向量
#基因差异的向量
genelist <- de_result$log2FoldChange
names(genelist) <- de_result$id
genelist <- sort(genelist,decreasing = T)

#构建本地库
#提取基因信息
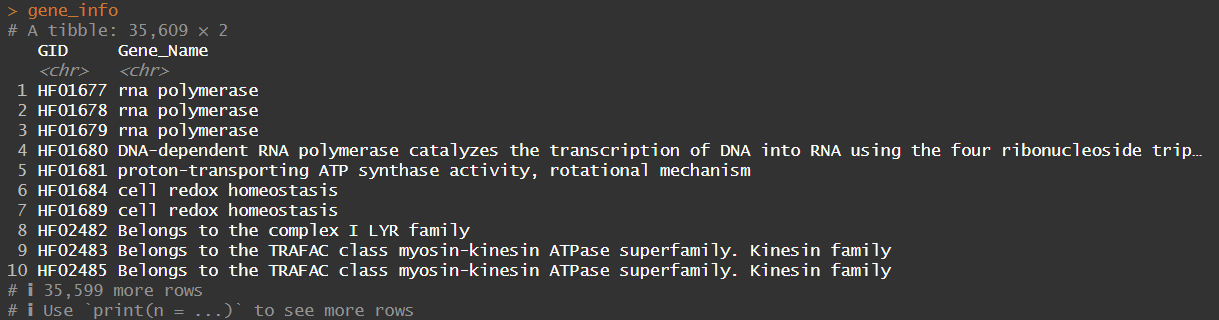
gene_info <- dplyr::select(emapper, GID , Gene_Name) %>%
dplyr::filter(!is.na(Gene_Name))
gene_info
#提取go信息
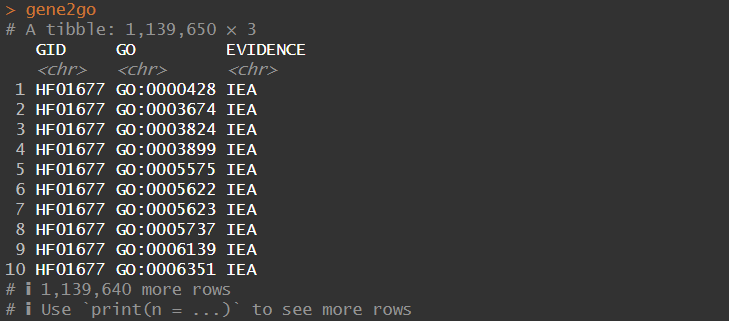
gene2go <- dplyr::select(emapper,GID,GO) %>%
separate_rows(GO,sep = ‘,’,convert = F) %>%
filter(!is.na(GO)) %>%
mutate(EVIDENCE = ‘IEA’)
gene2go
setwd(‘C:/Users/10395/Desktop/’) #甚至包的安装路径
#构建包
AnnotationForge::makeOrgPackage(gene_info = gene_info,
go = gene2go,
maintainer = ‘xiaoguo <pingguo @genek.tv>’,
author = ‘xiaoguo’,
outputDir = ‘./’,
tax_id = 0000,
genus = ‘X’,
species = ‘iaoguo’,
goTable = ‘go’,
version = ‘1.0’)

此时在桌面上会生成这个文件夹,然后我们再对该文件夹进行打包
#打包
pkgbuild::build(‘./org.Xiaoguo.eg.db’,dest_path = “C:/Users/10395/Desktop”)
![]()
随着打包的结束,你所选的文件夹内会生成以上的压缩文件,既是本地的安装R包
#新建文件夹
dir.create(‘R_Library’,recursive =T)
#安装包
install.packages(‘ C:/Users/10395/Desktop/org.Xiaoguo.eg.db_1.0.tar.gz’,
repos = NULL,#本地安装
lib = ‘R_Library’) #安装文件夹
#加载包
library(org.Xiaoguo.eg.db_1.0.tar.gz, lib = ‘R_Library’)
#富集分析
de_ego <- enrichGO(gene = gene,
OrgDb = org.Xiaoguo.eg.db,
keyType = ‘GID’,
ont = ‘BP’,
qvalueCutoff = 0.05,
pvalueCutoff = 0.05)
#将结果转为数据框模式
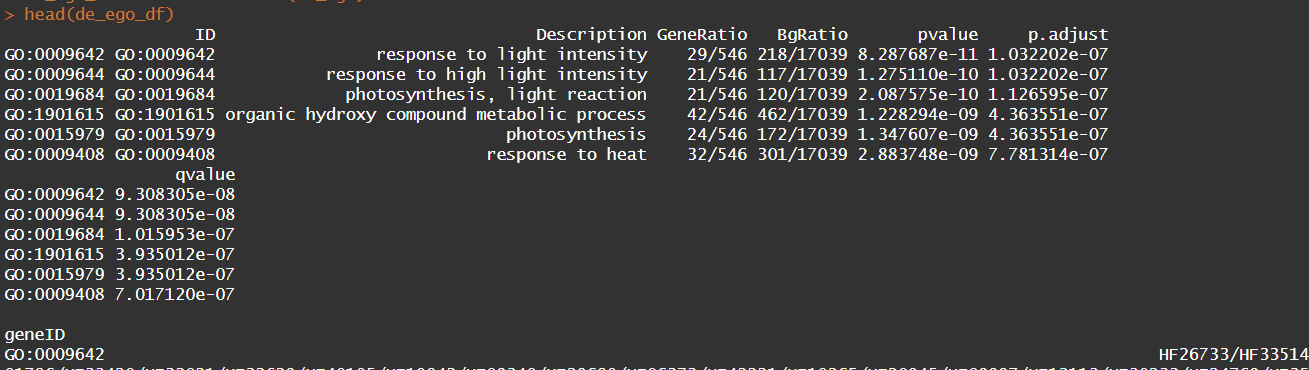
de_ego_df <- as.data.frame(de_ego)
head(de_ego_df)
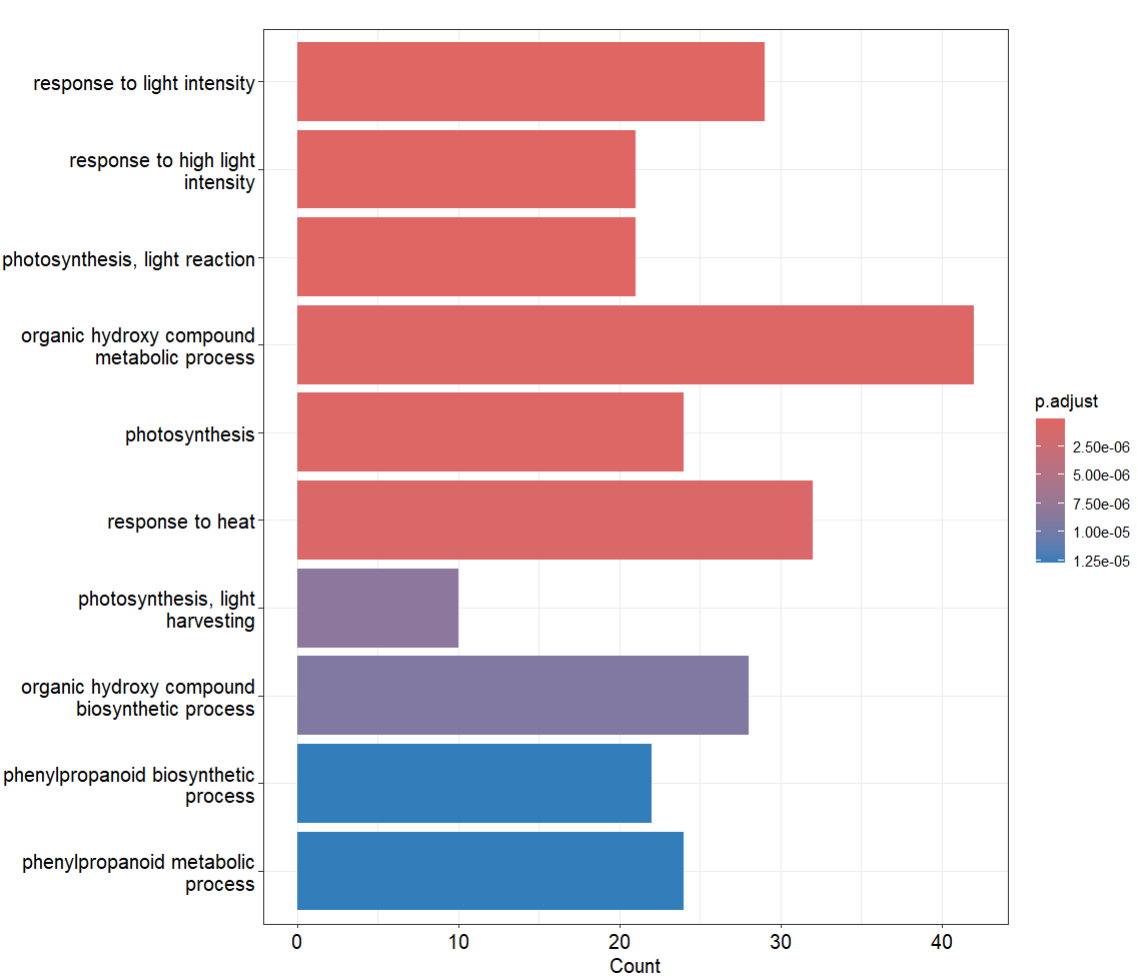
#可视化
P1 <- barplot(de_ego,showCategory = 10)
P1
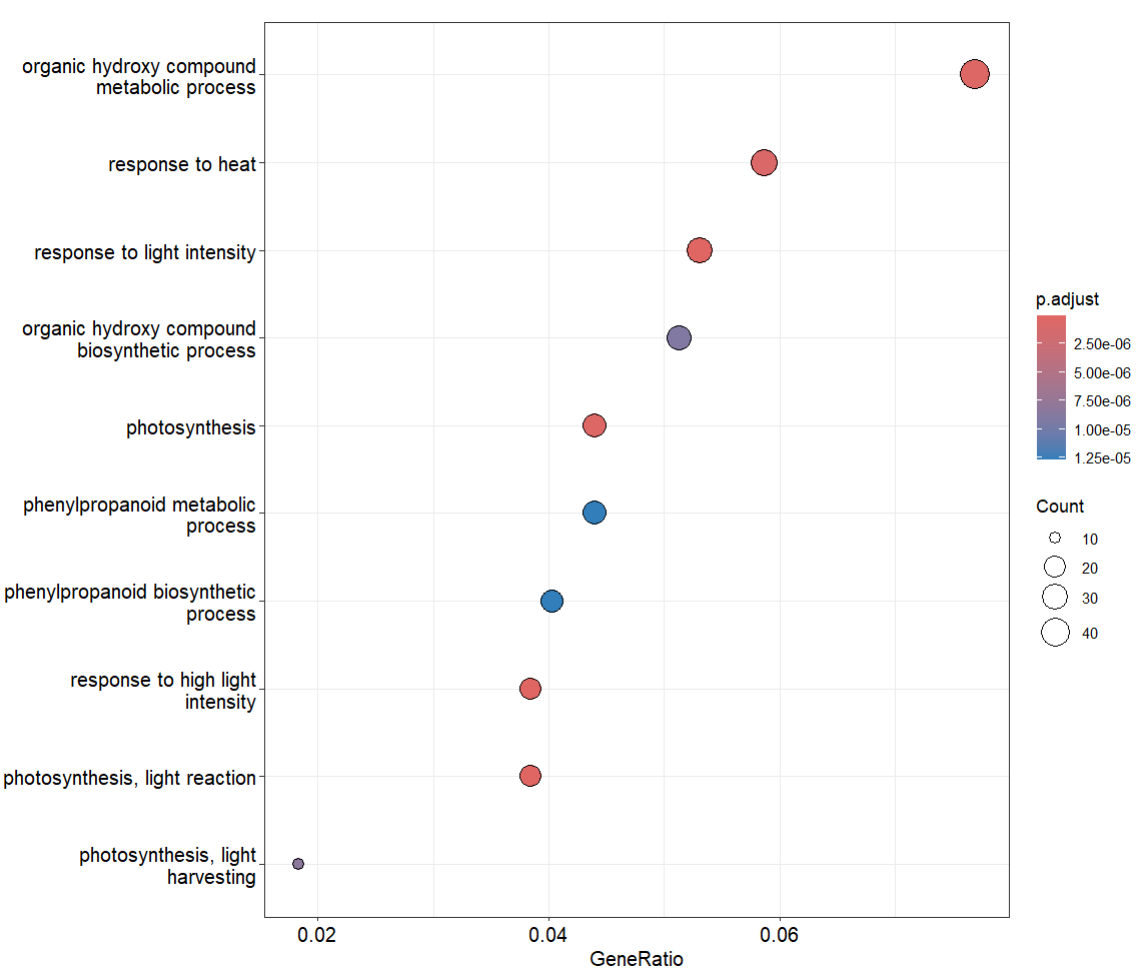
P2 <- enrichplot::dotplot(de_ego,showCategory = 10)
P2
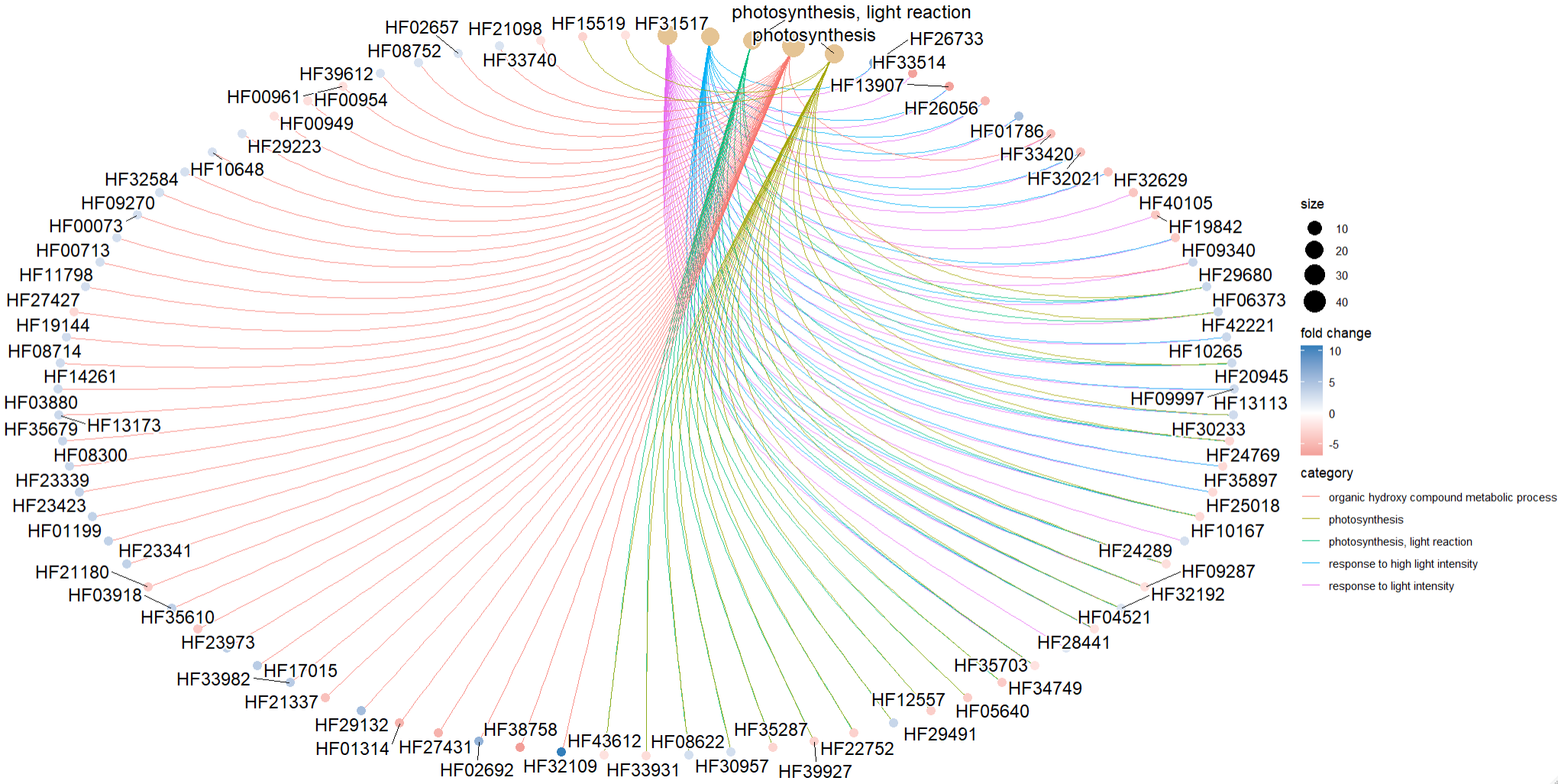
P3 <- cnetplot(de_ego,
foldChange = genelist,
showCategory = 5,
node_label = ‘all’ ,#category, gene all none
circular = TRUE,
colorEdge = TRUE)
P3
好啦,以上就是咱们无参转录组以及被eggNOG注释文件的富集分析结果哦。其实从步骤来看,的确不是很难,也便于理解,我们只需要通过已有文件将两个配置文件做好就OK了。但是,在这个过程中大家一定要注意两个事情,即我们配置文件的表头一定要按照我所写的来命名,因为在这个底层逻辑上,建库的包只认以该名字命名的列;另外就是的确有的数据不能被转换成数据框格式或转换的过程中以NULL结束进程,这是因为格式问题,这时候我们需要对他进行另外一种处理—用data.frame函数自己拼建数据框即结果文件:
result <- data.frame(de_ego@result[[“ID”]],
de_ego@result[[“Description”]],
de_ego@result[[“GeneRatio”]],
de_ego@result[[“BgRatio”]],
de_ego@result[[“pvalue”]],de_ego@result[[“p.adjust”]],
de_ego@result[[“qvalue”]],
de_ego@result[[“geneID”]],de_ego@result[[“Count”]])
这样的话也会生成和上述de_ego_df相同的数据框结果哦,也保证了各位小伙伴们忙碌了半天的成果不会付之东流。
另外,如果各位觉得直接获得的可视化结果不好看,这些函数本身还有多个参数可改变图形的样式而供小伙伴们使用哦。其次我们的确还可以利用现有的已经输出的结果文件再次进行二次绘图,相信肯定会比小果画的更好看呢!最后,如果各位小伙伴真的不想在数据可视化上花费太大时间,可以来小果的云生信平台哦,里面有大量成熟的可视化代码,只需要大家将数据输入就可以了哦,快来加入我们把!
http://www.biocloudservice.com/home.html