今天小果绘制一下分类变量变量热图,绘制该图需要三个输入文件,代码如下:
- 安装所需要的R包
BiocManager::install(“ComplexHeatmap”)
- 导入需要的R包
library(ComplexHeatmap)
- 导入数据
#差异表达分析中的logFC
logfc <- read.csv(“logFc.csv”, row.names = 1)
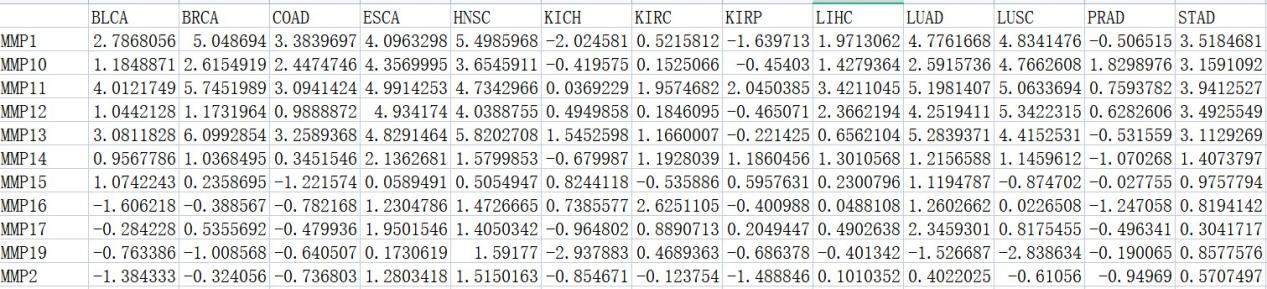
#差异分析中的P值
p.val <- read.csv(“PVal.csv”, row.names = 1)
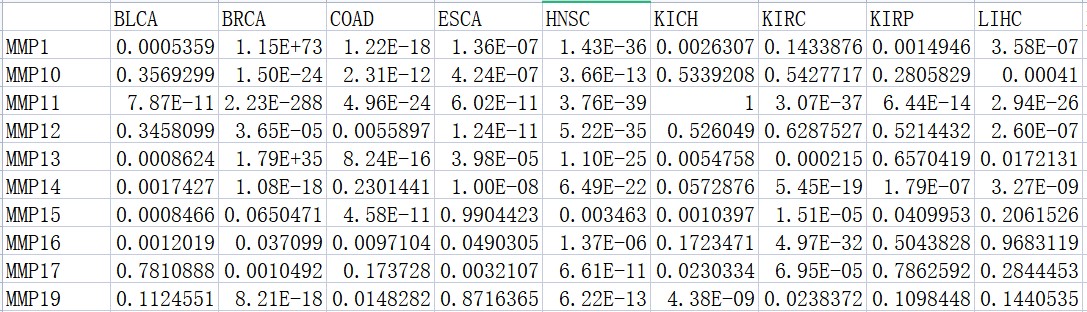
#基因分组
genetype <- read.csv(“genetype.csv”)
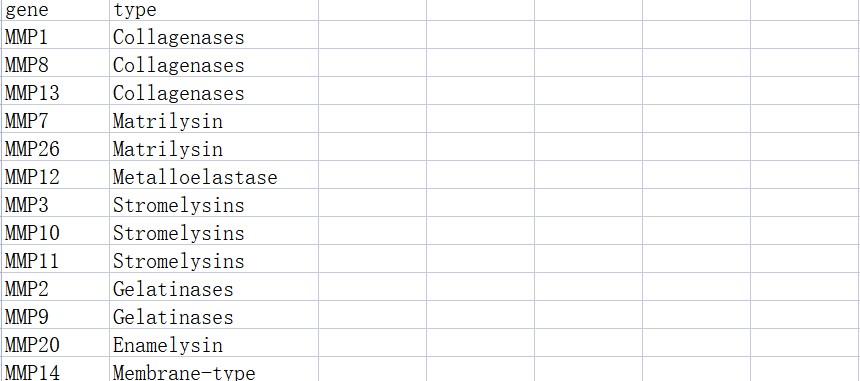
3.代码展示
#为确保热图基因的顺序和分组文件的顺序是一致的
#因此按照基因分组文件里的顺序,对logfc和p.val两个文件排序
logfcOrdered <- logfc[genetype$gene,]
p.valOrdered <- p.val[genetype$gene,]
## 对logfc进行分类
logfcCat <- apply(logfcOrdered, 2, function(x){
cut(x, breaks = c(-Inf, -2, -1, 1, 2, Inf),
labels = c(“< -2”, “-2 – -1”, “-1 – 1”, “1 – 2”, “> 2”))
})
rownames(logfcCat) <- rownames(logfcOrdered)
## 确保两个数据集的列名和行名顺序一致
if(!identical(rownames(logfcCat), rownames(p.valOrdered))) p.valOrdered <- p.valOrdered[rownames(logfcCat),]
if(!identical(colnames(logfcCat), colnames(p.valOrdered))) p.valOrdered <- p.valOrdered[colnames(logfcCat)]
## 把P> 0.05的数据标记为 P > 0.05
logfcCat[p.valOrdered >= 0.05] <- “P >= 0.05”
## 每个分类定义一个颜色
col_cat <- c(“> 2” = “#A80C3A”, “1 – 2” = “#ED5E57”, “-1 – 1” = “#DDD3D2”,
“-2 – -1” = “#6B9AB7”, “< -2” = “#2F5B89”, “P >= 0.05” = “white”)
cell_fun <- function(logfc, dataP, logfcCutoff = 1, PCutoff = 0.05,
darkcol = “black”, lightcol = “white”, digit = 2, fontsize = 6){
function(j, i, x, y, width, height, fill){
if(abs(logfc[i,j]) > logfcCutoff & dataP[i,j] < PCutoff){
grid.text(round(logfc, digit)[i, j], x, y,
gp = gpar(fontsize = fontsize, col = lightcol))
}else{
grid.text(round(logfc, digit)[i, j], x, y,
gp = gpar(fontsize = fontsize, col = darkcol))
}
}
}
## 定义注释信息
row_an <- HeatmapAnnotation(type = genetype$type, ##注释信息的内容
show_annotation_name = F, ## 是否显示注释的标题
col = list(type = an_col), ## 注释信息的颜色
show_legend = T, ## 是否显示注释信息的说明
annotation_legend_param = list(title = “MMP Subtype”), ## 注释信息图例的标题
which = “row”) #对行或者列进行注释
pdf(“rankHeatmap.pdf”, width = 8, height = 4)
Heatmap(matrix = logfcCat,
name = “logFC”, #主要图例的标题
rect_gp = gpar(col = “NA”, lwd = 1), #不画边框,或者用col = “grey”画灰色边框
col = col_cat, #热图颜色
row_names_side = “left”,
cell_fun = cell_fun(logfcOrdered, p.valOrdered),
row_names_gp = gpar(fontsize = 8), #基因名字号
column_names_gp = gpar(fontsize = 8), #肿瘤类型字号
column_names_rot = 45, #肿瘤类型呈45度
left_annotation = row_an) #左侧基因分类,如果不画,就筛掉这个参数
dev.off()
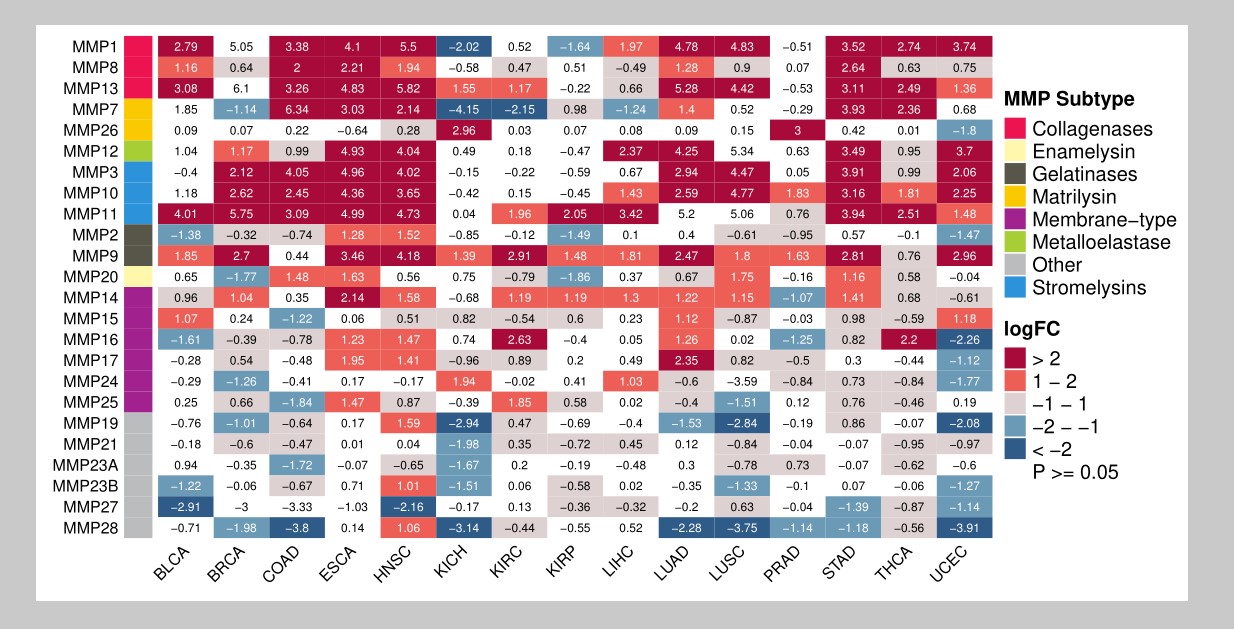
最终小果绘制出了分类变量热图,看起来效果不错,有需要的可以借鉴学习奥。