今天小果想带着小伙伴一起学习如何利用scMetabolism R包进行单细胞代谢活性的分析,该包可以对每一个细胞类型进行进行代谢活性的推断,使用的代谢数据集来自KEGG和REACTOME两个数据库,包含4种算法可以进行选择,ssGSEA,AUCell,VISION和GSVA,最后可以绘制气泡图,umap图和箱线图来展示不同类型细胞的代谢活性,想深入学习的小伙伴可以参考网站:https://github.com/wu-yc/scMetabolism,话不多说,开始今天的分享,代码如下:
- 安装需要的R包
install.packages(“devtools”)
install.packages(“data.table”)
install.packages(“wesanderson”)
install.packages(“AUCell”)
install.packages(“GSEABase”)
install.packages(“GSVA”)
install.packages(“ggplot2”)
install.packages(“rsvd”)
devtools::install_github(“YosefLab/VISION”)
devtools::install_github(“wu-yc/scMetabolism”)
- 导入需要的R包
library(phangorn)
library(scMetabolism)
library(ggplot2)
library(rsvd)
library(AUCell)
library(GSEABase)
library(GSVA)
- 示例数据
load(file = “pbmc_demo.rda”)
4.代码展示
#通过AUCell算法来计算细胞代谢活性(此处开始收费)
countexp.Seurat<-sc.metabolism.Seurat(obj = countexp.Seurat, method = “AUCell”, imputation =F, ncores = 2, metabolism.type = “KEGG”)
#obj表示单细胞seurat分析结果数据
#metho表示通过哪种算法进行计算
#ncores表示线程数
#metabolism.type表示选择的代谢数据库类型,其中KEGG数据库包含85条代谢途径,REACTOME数据库包含82条代谢途径。
#如果想提取metabolism score,可以运行以下命令。
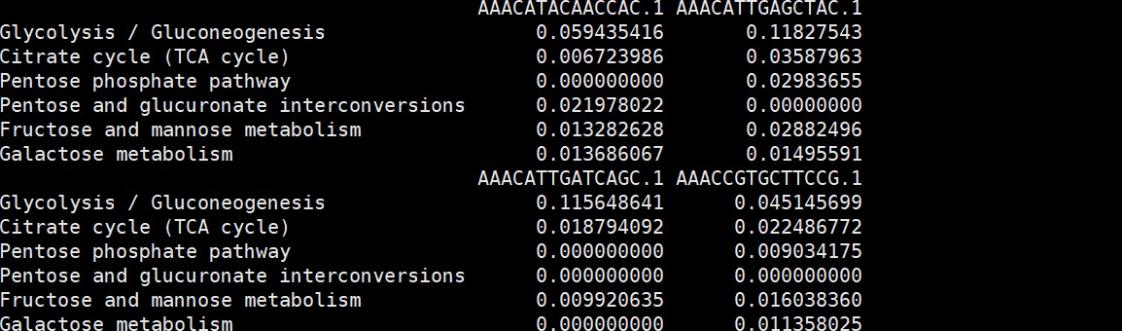
metabolism.matrix <- countexp.Seurat@assays$METABOLISM$score
metabolism.matrix 数据类型为一个矩阵。
#通过umap方式展示特定通路的细胞代谢活性
DimPlot.metabolism(obj = countexp.Seurat, pathway = “Glycolysis / Gluconeogenesis”, dimention.reduction.type = “umap”, dimention.reduction.run = F, size = 1)
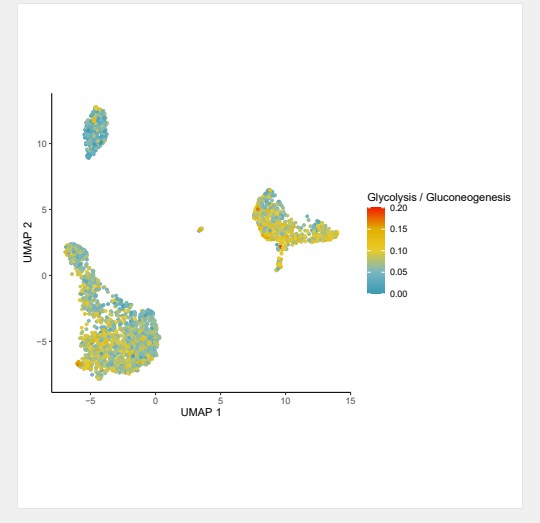
注:该图片通过umap的方式展示了特定代谢通路细胞的代谢活性,通过不同颜色来表示代谢活性的高低。
#通过气泡图的方式展示细胞代谢活性
input.pathway<-c(“Glycolysis / Gluconeogenesis”, “Oxidative phosphorylation”, “Citrate cycle (TCA cycle)”)
DotPlot.metabolism(obj = countexp.Seurat, pathway = input.pathway, phenotype = “ident”, norm = “y”)
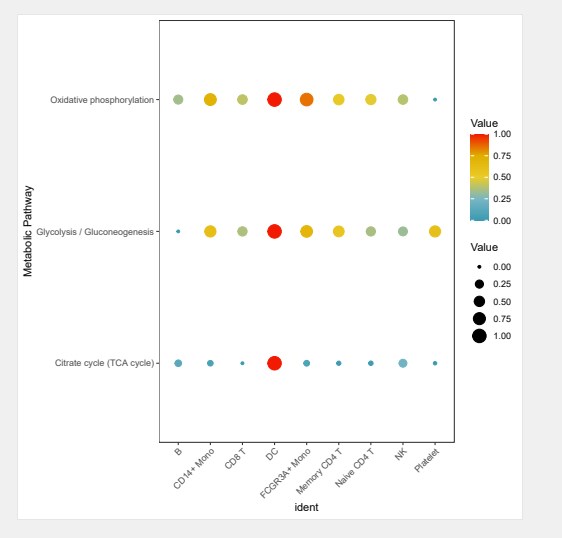
注:该图表示不同细胞的活性的代谢活性,横坐标表示细胞类型,纵坐标表示代谢途径,颜色代表value值,颜色越深表示活性越高。
#通过箱线图的方式展示细胞代谢活性
BoxPlot.metabolism(obj = countexp.Seurat, pathway = input.pathway, phenotype = “ident”, ncol = 1)
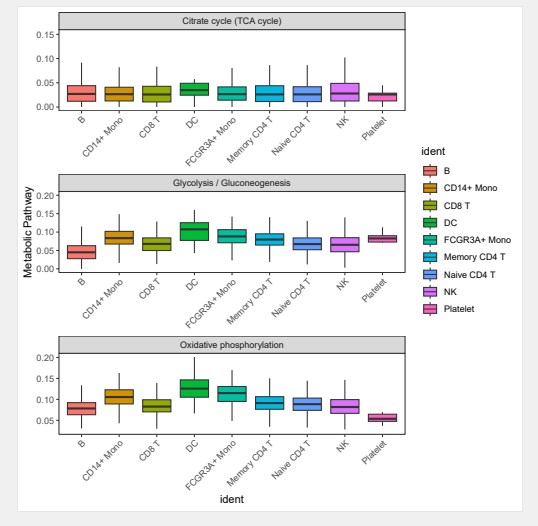
注:该图通过分面箱线图来展示不同代谢通路细胞的代谢活性,纵坐标表示细胞活性高低,横坐标表示细胞类型。
#如果数据只有UMI count ,可以通过以下方式进行分析,需要注意的是数据类型为列为细胞ID,行为基因name。
metabolism.matrix<-sc.metabolism(countexp = countexp, method = “AUCell”, imputation = F, ncores = 2, metabolism.type = “KEGG”)
最终小果顺利的完成了单细胞代谢活性分析,整个分析过程还是很easy的,小伙伴们有需要的可以借鉴学习奥,在做该分析时还是需要对单细胞数据分析要熟练掌握,如果小伙伴需要进行单细胞数据分析,推荐大家尝试本公司新开发的云平台生信分析小工具,零代码完成分析,云平台网址:http://www.biocloudservice.com/home.html,包含单细胞分析(http://www.biocloudservice.com/366/366.php),绘制单细胞tSNE图(http://www.biocloudservice.com/229/229.php),单细胞数据绘制小琴图(http://www.biocloudservice.com/788/788.php)等小工具,小果今天的分享就到这里,欢迎大家和小果一起讨论学习奥,下期再见。