单细胞内置数据处理和分析
今天小果想学习分享一下单细胞内置数据的处理和分析过程,提高大家对公共数据的利用效率,有需要的可以学习掌握一下奥,代码如下:
- 安装需要的R包
install.packages(“BiocManager”)
BiocManager::install(“Seurat”)
devtools::install_github(‘satijalab/seurat-data’,force = TRUE)
install.packages(“patchwork”)
- 导入需要的R包
library(Seurat)
library(SeuratData)
Library(patchwork)
- 代码展示
InstallData(“ifnb”)
LoadData(“ifnb”)
# ifnb数据集中包含两个样本数据,分别为CTRL和STIM
head(ifnb@meta.data)
table(ifnb@meta.data$stim)
# 数据拆分成两组(STIM和CTRL)
# 使用splitobject将ifnb数据集分割成两个seurat对象的列表(STIM和CTRL)
ifnb.list <- SplitObject(ifnb, split.by = “stim”)
# 对两个数据集单独进行normalize并识别各自的高变异基因
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = “vst”, nfeatures = 2000)
})
# 计算两个数据中排完rank后top2000的高变异基因
features <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 2000)
# 使用上述高变异基因在两个基因集中分别执行PCA
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- ScaleData(x, features = features, verbose = T)
x <- RunPCA(x, features = features, verbose = T)
})
## 对两个数据集进行整合
# 首先使用findinintegrationanchors函数标识锚点,然后使用IntegrateData函数将两数据集整合在一起
# 可以通过调整k.anchor参数调整两个样本整合的强度,默认为5,数字越大整合强度越强
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features, reduction = “rpca”, k.anchor = 20)
immune<- IntegrateData(anchorset = immune.anchors)
# 常规流程,scale数据,跑PCA,使用前30个PC进行降维跑UMAP,计算Neighbors,分cluster
immune <- ScaleData(immune, verbose = FALSE)
immune <- RunPCA(immune, npcs = 30, verbose = FALSE)
immune <- RunUMAP(immune, reduction = “pca”, dims = 1:30)
immune <- FindNeighbors(immune, reduction = “pca”, dims = 1:30)
immune<- FindClusters(immune, resolution = 0.5)
immune@meta.data
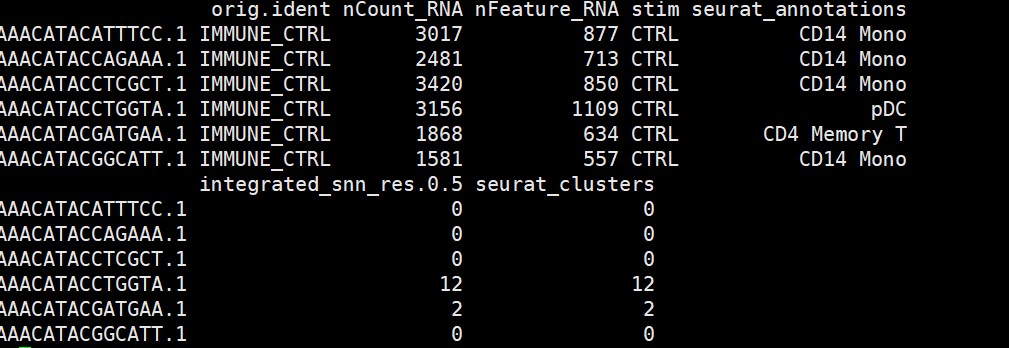
# 可视化
p <- DimPlot(immune, reduction = “umap”, group.by = “stim”)
p21<- DimPlot(immune, reduction = “umap”, group.by = “seurat_annotations”,label = TRUE,repel = TRUE)
#拼图
pdf(“immune.UMAP.pdf”, height = 5,width = 12)
p+ p1
dev.off()
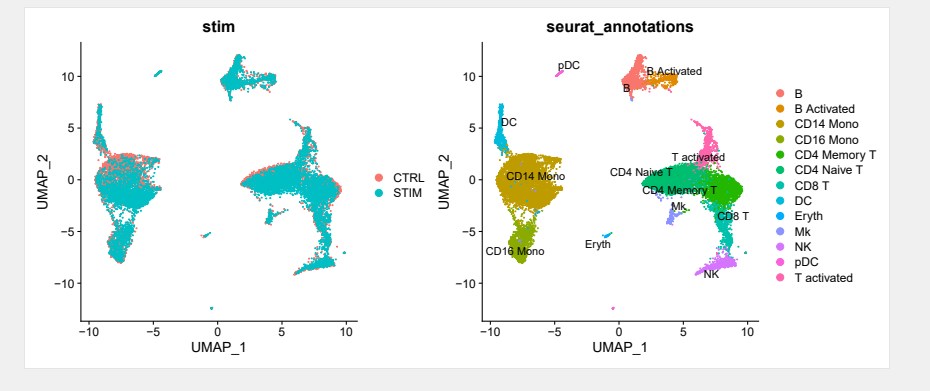
最终我们对内置数据集ifnb进行了数据处理,其中处理过程包含了数据集的整合和拆分等过程,并进行了单细胞常规流程分析,该分析所需要的R包本公司服务器已经全部安装,有需要的小伙伴可以联系小果奥,今天小果的分享就到这里。
今天小果想学习分享一下单细胞内置数据的处理和分析过程,提高大家对公共数据的利用效率,有需要的可以学习掌握一下奥,代码如下:
- 安装需要的R包
install.packages(“BiocManager”)
BiocManager::install(“Seurat”)
devtools::install_github(‘satijalab/seurat-data’,force = TRUE)
install.packages(“patchwork”)
- 导入需要的R包
library(Seurat)
library(SeuratData)
Library(patchwork)
- 代码展示
InstallData(“ifnb”)
LoadData(“ifnb”)
# ifnb数据集中包含两个样本数据,分别为CTRL和STIM
head(ifnb@meta.data)
table(ifnb@meta.data$stim)
# 数据拆分成两组(STIM和CTRL)
# 使用splitobject将ifnb数据集分割成两个seurat对象的列表(STIM和CTRL)
ifnb.list <- SplitObject(ifnb, split.by = “stim”)
# 对两个数据集单独进行normalize并识别各自的高变异基因
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = “vst”, nfeatures = 2000)
})
# 计算两个数据中排完rank后top2000的高变异基因
features <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 2000)
# 使用上述高变异基因在两个基因集中分别执行PCA
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- ScaleData(x, features = features, verbose = T)
x <- RunPCA(x, features = features, verbose = T)
})
## 对两个数据集进行整合
# 首先使用findinintegrationanchors函数标识锚点,然后使用IntegrateData函数将两数据集整合在一起
# 可以通过调整k.anchor参数调整两个样本整合的强度,默认为5,数字越大整合强度越强
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features, reduction = “rpca”, k.anchor = 20)
immune<- IntegrateData(anchorset = immune.anchors)
# 常规流程,scale数据,跑PCA,使用前30个PC进行降维跑UMAP,计算Neighbors,分cluster
immune <- ScaleData(immune, verbose = FALSE)
immune <- RunPCA(immune, npcs = 30, verbose = FALSE)
immune <- RunUMAP(immune, reduction = “pca”, dims = 1:30)
immune <- FindNeighbors(immune, reduction = “pca”, dims = 1:30)
immune<- FindClusters(immune, resolution = 0.5)
immune@meta.data
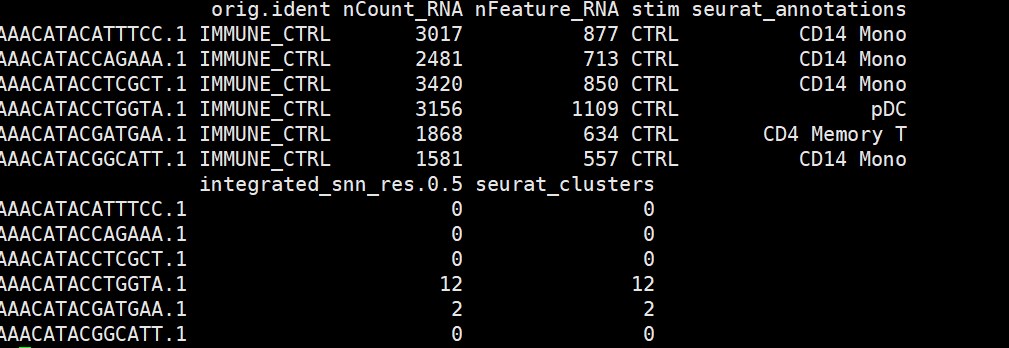
# 可视化
p <- DimPlot(immune, reduction = “umap”, group.by = “stim”)
p21<- DimPlot(immune, reduction = “umap”, group.by = “seurat_annotations”,label = TRUE,repel = TRUE)
#拼图
pdf(“immune.UMAP.pdf”, height = 5,width = 12)
p+ p1
dev.off()
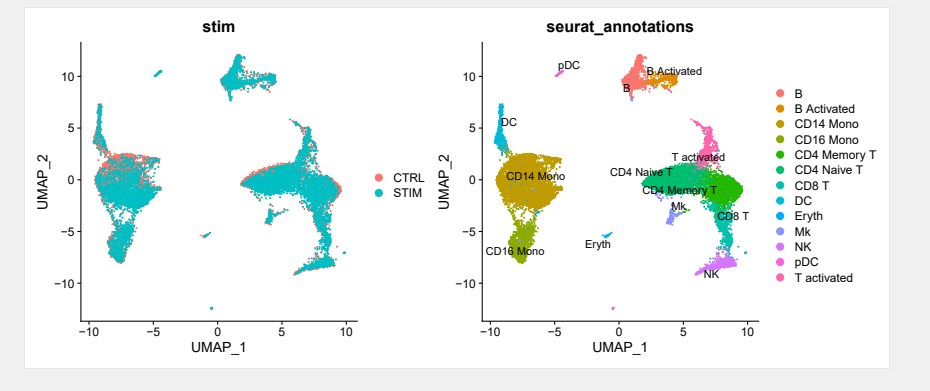
最终我们对内置数据集ifnb进行了数据处理,其中处理过程包含了数据集的整合和拆分等过程,并进行了单细胞常规流程分析,该分析所需要的R包本公司服务器已经全部安装,有需要的小伙伴可以联系小果奥,今天小果的分享就到这里。