单细胞分析在现在非常火,今天小果也想带着大家开始学习梳理一下单细胞分析流程,简单熟悉一下单细胞分析所包含的内容,分析内容如下:
- 安装单细胞分析R包
install.packages(“BiocManager”)
BiocManager::install(“Seurat”)
library(Seurat)
- 单细胞测序文件的读入和Seurat对象构建
#读取数据所在的文件夹-标准的10X文件
pbmc.data <- Read10X(data.dir = “filtered_gene_bc_matrices/hg19/”)
#创建Seurat文件
pbmc <- CreateSeuratObject(counts = pbmc.data, project = “pbmc3k”, min.cells = 3, min.features = 200)
min.cells表示筛选至少在3个细胞中表达的基因
min.features表示筛选至少有200个基因表达的细胞
- 单细胞数据质控(QC)
#先计算线粒体比例
pbmc[[“percent.mt”]] <- PercentageFeatureSet(pbmc, pattern = “^MT-“)
#用小提琴图展示控前指标
VlnPlot(pbmc, features = c(“nFeature_RNA”, “nCount_RNA”, “percent.mt”), ncol = 3,group.by=”orig.ident”,pt.size=0))
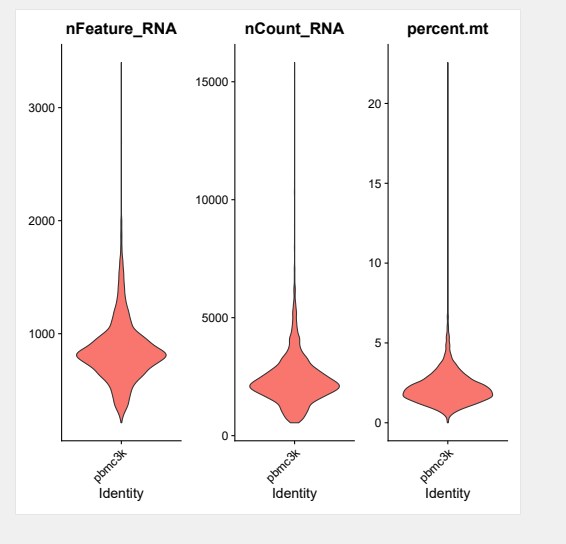
#进行质控(可以根据自己的数据进行过滤)
pbmc<-subset(pbmc,nFeature_RNA>200 &nFeature_RNA<5000 &percent.mt <15)
VlnPlot(pbmc, features = c(“nFeature_RNA”, “nCount_RNA”, “percent.mt”), ncol = 3,group.by=”orig.ident”,pt.size=0)
- 单细胞数据标准化及计算高变基因
#数据标准化
pbmc <- NormalizeData(pbmc, normalization.method = “LogNormalize”, scale.factor = 10000)
用log标准化等方式降低其他干扰因素的影响
#计算高变异基因
pbmc <- FindVariableFeatures(pbmc, selection.method = “vst”, nfeatures = 2000)
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
- 单细胞数据UMAP降维聚类
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
#细胞聚类
pbmc <- FindNeighbors(pbmc, dims = 1:10)
#计算SNN
pbmc <- FindClusters(pbmc, resolution = 0.5)
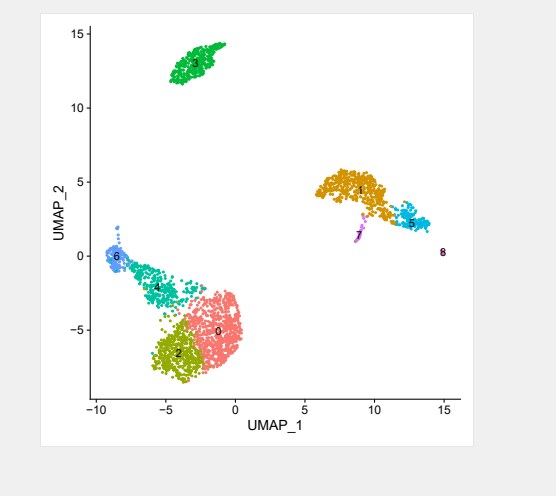
#UMAP非线性降维分析
pbmc <- RunUMAP(pbmc, dims = 1:10)
#绘制UMAP降维聚类图
DimPlot(pbmc, reduction = “umap”, label=T)+ NoLegend()
- 单细胞数据marker基因鉴定
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
signifcant.markers<-pbmc.markers[pbmc.markers$p_val_adj<0.2,]
#保存marker基因数据
write.csv(signifcant.markers,file=”signifcant.markers.csv”)
#marker基因可视化
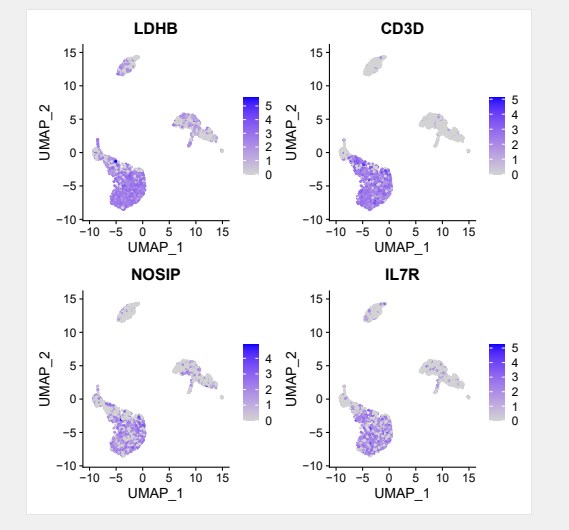
#可以用自己感兴趣的marker基因进行展示
markers<-c(“LDHB”,“CD3D”,”NOSIP”,”IL7R”)
##UMAP图
FeaturePlot(pbmc,features=markers)
##点图
DotPlot(pbmc,features=markers)+coord_flip()
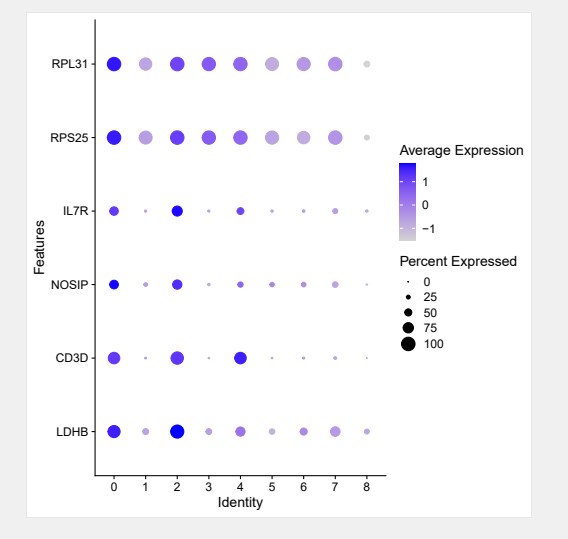
##热图
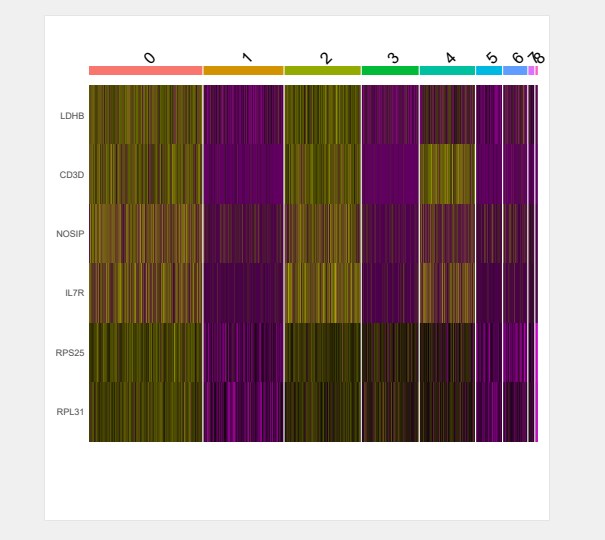
- 单细胞数据细胞群注释
# 给每个cluster标注细胞类型(一般细胞群的注释通过人工查文献获得的注释结果比较准确)
new.cluster.ids <- c(“T”, “Mono”, “T”, “B”, “T”, “Mono”, “NK”, “DC”, “Platelet”)
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
# 添加细胞类型至meta data
pbmc@meta.data$celltype <- pbmc@active.ident
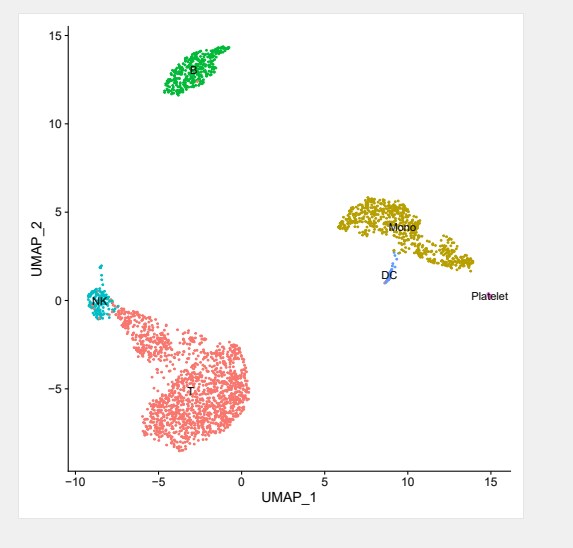
哈哈,这就是单细胞数据分析的基本流程,一般单细胞数据分析都需要很大的计算资源奥,通常都会利用高性能服务器来进行数据分析,小果推荐大家使用本公司的服务器,单细胞分析所需要的软件和R包都已安装好,有需要的可以联系小果,本期的分享就到这里。