今天小果带大家一起来认识单细胞测序的另一个常用工具Monocle,那就和我一起往下学习吧!~~
什么是Monocle?
在学习一个工具之前,我们首先要大概了解这个工具的基本来源和用途哦,这是每一个生信人都要养成的良好习惯!那就和小果一起来看看到底什么是Monocle吧!
其实,Monocle包可以根据每个细胞在学习轨迹上的进展对其进行排序,这一功能我们可以联想一些排序算法、排序工具等等哦。但是!要注意的是Monocle不是跟踪表达式随时间变化的函数,而是跟踪沿轨迹变化的函数,这样的变化表面看起来和时间变化有关却又不是时间,我们也就称之为伪时间,小果这样讲是不是就通俗易懂hen多了呢?
Monocle有几大功能模块?
众所周知,Monocle包也是单细胞测序中常用到的包之一。
那么Monocle可以帮助我们进行哪些方面的单细胞分析呢?和小果一起来看看吧!
1.聚类,分类,细胞计数。
2.细胞的差异表达分析。
3.构建单细胞轨迹。即在细胞发育、疾病以及整个生命过程里,细胞从一个状态转成另一个状态。
接下来,让小果带你学习如何安装Monocle包以及如何简单地使用Monocle包来对单细胞数据进行质控吧!!
Monocle包的安装
monocle包的安装有好几种方式,接下来是小果整理的三种安装monocle包的方法,快来和小编一起学习吧!~
- 通过Bioconducor安装
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
#下载monocle依赖的其它包
BiocManager::install(c(‘BiocGenerics’, ‘DelayedArray’, ‘DelayedMatrixStats’,
‘limma’, ‘lme4’, ‘S4Vectors’, ‘SingleCellExperiment’,
‘SummarizedExperiment’, ‘batchelor’, ‘HDF5Array’,
‘terra’, ‘ggrastr’))
BiocManager::install(“monocle”)
- dyno安装
# install.packages(“devtools”)
devtools::install_github(“dynverse/dyno”)
- 官网安装
source(“http://bioconductor.org/biocLite.R”)
biocLite()
biocLite(“monocle”)
以上三种方法,小果最推荐的还是第一种,小果在下载的时候用的也是第一种方法哦~快速简单且高效,同学们根据自身情况自行参考哈!~
Monocle对单细胞数据进行质控
安装成功后,我们和小果一起来看一下这个包的简单使用方法吧!
- step1:准备需要的R包
对于我们本次学习要用的数据从哪里来呢?不要急!小果给大家准备了两个R包来帮助我们进行后续的工作!让我们一起来看一下!
本次质控分析我们使用scRNAseq中的数据作为基础数据,并用来构建后续的数据矩阵,所以也需要我们提前下载好scRNAseq数据包哦!
library(monocle)
library(scRNAseq)
library(dplyr)
- step2:准备数据
小果这次使用的是scRNAseq里面的示例数据fluidigm哦,同学们自行选择哦!!。
虽然这个示例数据本身是一个数据对象,但我们还需从中提取出后续质控所需要的部分,再组成新的对象。 简单来说,就是我们需要对数据进行“二次包装”,转为我们需要的数据格式哦~
fluidigm = ReprocessedFluidigmData()
 提示信息如下:
提示信息如下:
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)

dim(ct)
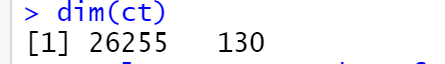
我们可以看到,在我们创建的数据中有130个细胞,26255个基因哦。
- step3:构建数据对象
gene_ann <- data.frame(
gene_short_name = row.names(ct),
row.names = row.names(ct)
)
pd <- new(“AnnotatedDataFrame”,
data=sample_ann)
fd <- new(“AnnotatedDataFrame”,
data=gene_ann)
cds <- newCellDataSet(
ct,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
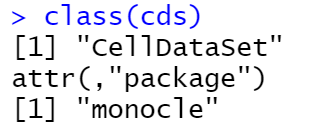
class(cds)
- step4:数据质控
哈哈,又到了小果最喜欢的数据质控环节啦!首先我们在数据质控之前一起来看一下数据过滤前的数据维度吧!
可以看到目前的数据维度是130个细胞,26255个基因。
dim(cds)
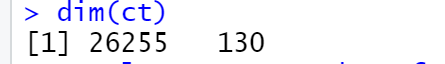
cds <- detectGenes(cds, min_expr = 0.1)
head(fData(cds))

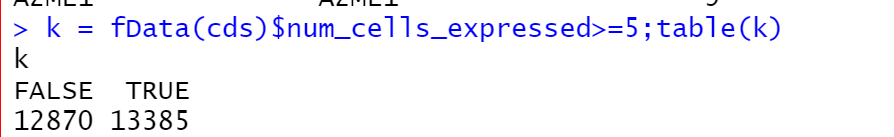
k = fData(cds)$num_cells_expressed>=5;table(k)
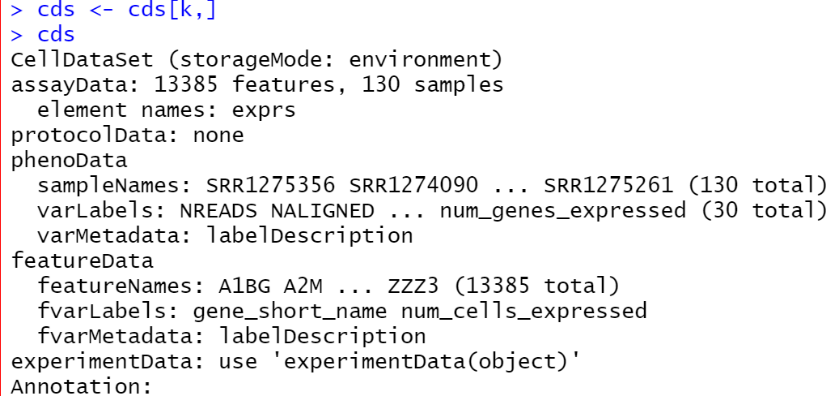
cds <- cds[k,]
cds
dim(cds)
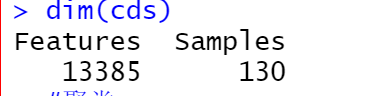
现在,再和小果一起来看一下过滤后的基因维度吧!!
过滤基因的后是:13385 features, 130 samples,很低的标准也过滤掉了一半的基因。
D=====( ̄▽ ̄*)b·········
今天的小果课堂就告一段落啦!
以上小果的一系列操作就是如何安装单细胞测序Monocle包和对单细胞测序数据进行质控过滤的基本流程,你们学会了吗!更多Monocle使用方法也请参考Monocle官方网站和帮助文档哦!~
还有什么别的问题欢迎大家来给小果积极留言,小果都会尽力帮助大家!~~