AUCell“识别”活性基因集
各位同学们!小果技能课堂终于又双缀开课啦!今天我们要学习一个全新的单细胞测序工具来解决一个特定的问题:
通过什么样的方法就能鉴别出细胞中存在的“活性”基因集并将它可视化展示呢?不要急,小果带你一起探讨!
AUCell安装
没错,我们今天要用到的就是R包—–AUCell,AUCell具有强大的识别功能和绘图功能,能够轻松解决我们现在想解决的问题哦。那就先和小果一起来完成AUCell包的安装吧!
BiocManager::install(“AUCell”,force = TRUE)
#本次实验还要用到的相关依赖
#我们需要用GEOquery来导入我们后续需要用到的矩阵数据
BiocManager::install(“GEOquery”,force = TRUE)
准备数据
在AUCell进行识别并输出之前,小果要准备最基本的数据,对于本次实验,我们用到的数据有两种,一种是“矩阵数据”,一种是“基因数据”哦 ,使用之前下载好的“GEOquery”包就可以直接在R中下载ncbi上的数据集。我们在下载好后,通过R中对数据处理的函数将其进行二次封装,从而将其转化为数据矩阵的形式~
- 准备矩阵数据
library(GEOquery)
library(Matrix)
gsdata <- getGEO(‘GSE60361’) # does not work, the matrix is in a suppl file
geo_file <- “GSE60361_C1-3005-Expression.txt.gz”
download.file(‘https://ftp.ncbi.nlm.nih.gov/geo/series/GSE60nnn/GSE60361/suppl/GSE60361_C1-3005-Expression.txt.gz’, destfile = geoFile)
#数据封装
eprMtrx <- fread(geoFile, sep = “\t”)
gen_name <- unname(unlist(eprMtrx[, 1, with = FALSE]))
eprMtrx <- as.matrix(exprMatrix[, -1, with = FALSE])
rownames(eprMtrx) <- gen_name
exprMatrix <- exprMatrix[unique(rownames(exprMatrix)), ]
dim(exprMatrix)
eprMtrx <- as(eprMtrx, “dgCMatrix”)
#数据保存
mouseExprMatrix <- eprMtrx
save(mouseExprMatrix, file = “exprMatrix.RData”)
- 准备基因数据
对于基因集数据,我们可以直接从AUCell软件包中自带的数据集直接调取,是不是很方便呢!让小果带大家来具体看看怎么操作吧!
library(AUCell)
library(GSEABase)
gmtf <- paste(file.path(system.file(“examples”, package = “AUCell”)), “geneSignatures.gmt”,
sep = “/”)
gene-Sets <- getGmt(gmtf)
gene-Sets <- subsetGeneSets(gene-Sets, rownames(eprMtrx))
cbind(nGenes(gene-Sets))
gene-Sets <- setGeneSetNames(gene-Sets, newNames = paste(names(geneSets), ” (“, nGenes(geneSets),
“g)”, sep = “”))
set.seed(321)
extraGeneSets <- c(GeneSet(sample(rownames(exprMatrix), 50), setName = “Random (50g)”),
GeneSet(sample(rownames(exprMatrix), 500), setName = “Random (500g)”))
countsGene <- apply(exprMatrix, 1, function(x) sum(x > 0))
extraSets <- c(extraSets, GeneSet(sample(names(countsGene)[which(countsGene >
quantile(countsGene, probs = 0.95))], 100), setName = “HK-like (100g)”))
gene-Sets <- GeneSetCollection(c(gene-Sets, extraSets))
names(geneSets)
- 识别细胞中的活性基因集
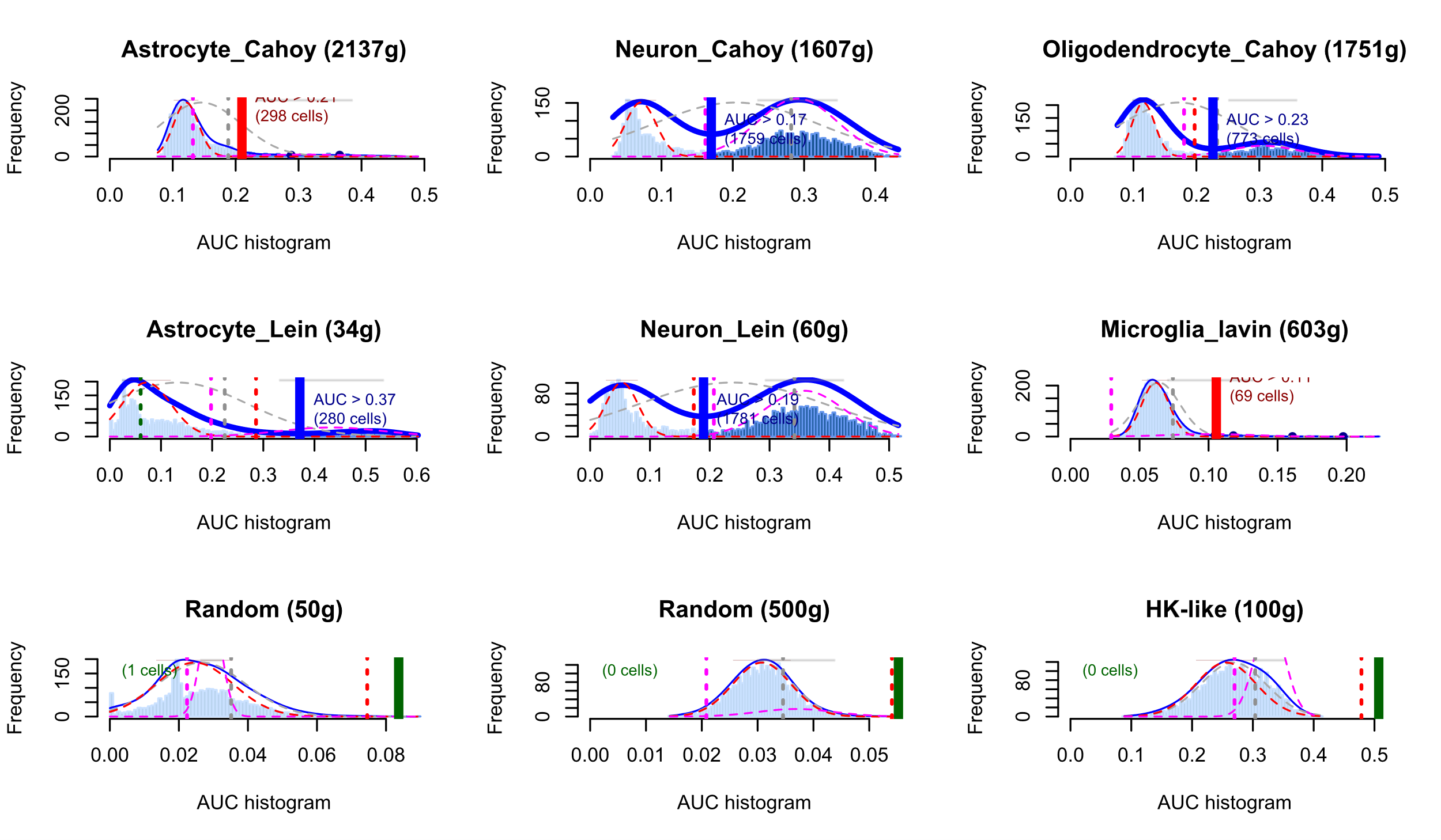
当AUCell识别了细胞中的活性基因后,会根据活性基因在细胞中表达的情况用曲面面积来进行表示哦!调用AUCell的可视化函数,我们也可以直观的看到数据集中的细胞中表达出的活性强度,从而也有助于我们从数据群中找到活性强的一系列细胞!接下来小果带大家一起看看AUCell的强大绘图功能吧!!
set.seed(120)
par(mfrow = c(3, 3))
#将矩阵数据和基因数据作为输入
cells_AUC <- AUCell_run(eprMtrx, gene-Sets)
cells <- AUCell_exploreThresholds(cells_AUC, plotHist = TRUE, assign = TRUE)
来和小果一起看看输出结果吧!注意,这里因为绘制的图很多,所以我们需要将右下角的图框拉大一些哦!不然可能会导致看不到绘图结果哦!!
D=====( ̄▽ ̄*)b========================
好啦,今天这个技能你有好好跟小果一起学嘛?不知道你学会了没有捏,更多小技能请多多关注小果的公众号哦!!