今天小果想为大家安利一个R包ClusterGVis,该包由老俊俊开发,前期该包可以进行Bluk-seq表达矩阵的处理,可以同时绘制聚类+分组表达趋势折线图+功能注释的组合图,通过一张热图可以了解差异基因可以划分成几个cluster,每个cluster的表达随着时间是如何变化,以及这些cluster变化的基因通过GO或者KEGG功能注释了解其功能;最近看到该包可以对接单细胞数据了,增加了 prepareDataFromscRNA 函数来整理准备单细胞的数据,可以绘制出像Bluk-seq一样的组合图,效果非常不错,小果接下来就和大家一起学习该包的用法,如何对接单细胞数据,话不多说开始今天的分享,代码如下:
- 安装需要的R包
#重新安装获得此功能,如果以前没有安装可以忽略,直接安装就可以
install.packages(“devtools”)
devtools::install_github(“junjunlab/ClusterGVis”)
BiocManager::install(“org.Hs.eg.db”)
install.packages(“ggplot2”)
- 导入需要的R包
library(ClusterGVis)
library(org.Hs.eg.db)
library(ggplot2)
- 代码展示
#导入单细胞上游分析结果文件
load(“pbmc.rda”)
#单细胞数据细胞群进行注释
new.cluster.ids <- c(“Naive CD4 T”, “CD14+ Mono”, “Memory CD4 T”, “B”, “CD8 T”, “FCGR3A+ Mono”,”NK”, “DC”, “Platelet”)
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
#寻找marker基因
pbmc.markers.all <- Seurat::FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
#挑选表达量前十的marker基因
pbmc.markers <- pbmc.markers.all %>%
dplyr::group_by(cluster) %>%
dplyr::top_n(n = 20, wt = avg_log2FC)
#利用prepareDataFromscRNA函数准备数据,showAverage 参数设为 TRUE 则表示对 基因细胞亚群一样的细胞取均值进行绘图,否则就是所有细胞进行绘图,默认使用 seurat 对象的 RNA assay 的 data 数据。
st.data <- prepareDataFromscRNA(object = pbmc,
diffData = pbmc.markers,
showAverage = TRUE)
#对每个cluster进行富集分析,这里采用GO富集分析,有需要的可以选择kegg富集分析
enrich <- enrichCluster(object = st.data,
OrgDb = org.Hs.eg.db,
type = “BP”,
organism = “hsa”,
pvalueCutoff = 0.5,
topn = 5,
seed = 5201314)
#挑选需要展示的marker基因
markGenes = unique(pbmc.markers$gene)[sample(1:length(unique(pbmc.markers$gene)),40,
replace = F)]
#绘制cluster基因表达折线图
visCluster(object = st.data,
plot.type = “line”)
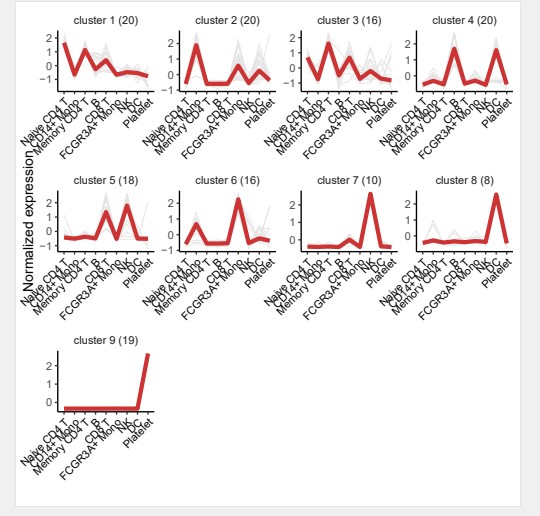
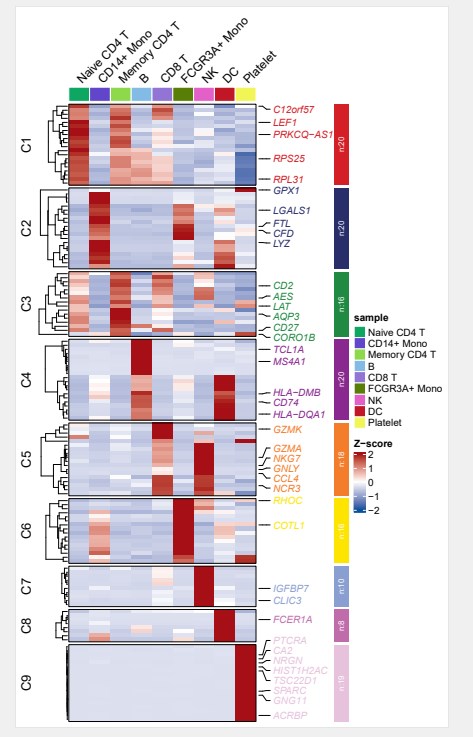
#绘制热图
pdf(‘sc1.pdf’,height = 10,width = 6,onefile = F)
visCluster(object = st.data,
plot.type = “heatmap”,
column_names_rot = 45,
markGenes = markGenes,
cluster.order = c(1:9))
dev.off()
#绘制热图并添加富集注释和分组折线图
pdf(‘sc2.pdf’,height = 10,width = 14,onefile = F)
visCluster(object = st.data,
plot.type = “both”,
column_names_rot = 45,
show_row_dend = F,
markGenes = markGenes,
markGenes.side = “left”,
annoTerm.data = enrich,
line.side = “left”,
cluster.order = c(1:9),
go.col = rep(jjAnno::useMyCol(“stallion”,n = 9),each = 5),
add.bar = T)
dev.off()
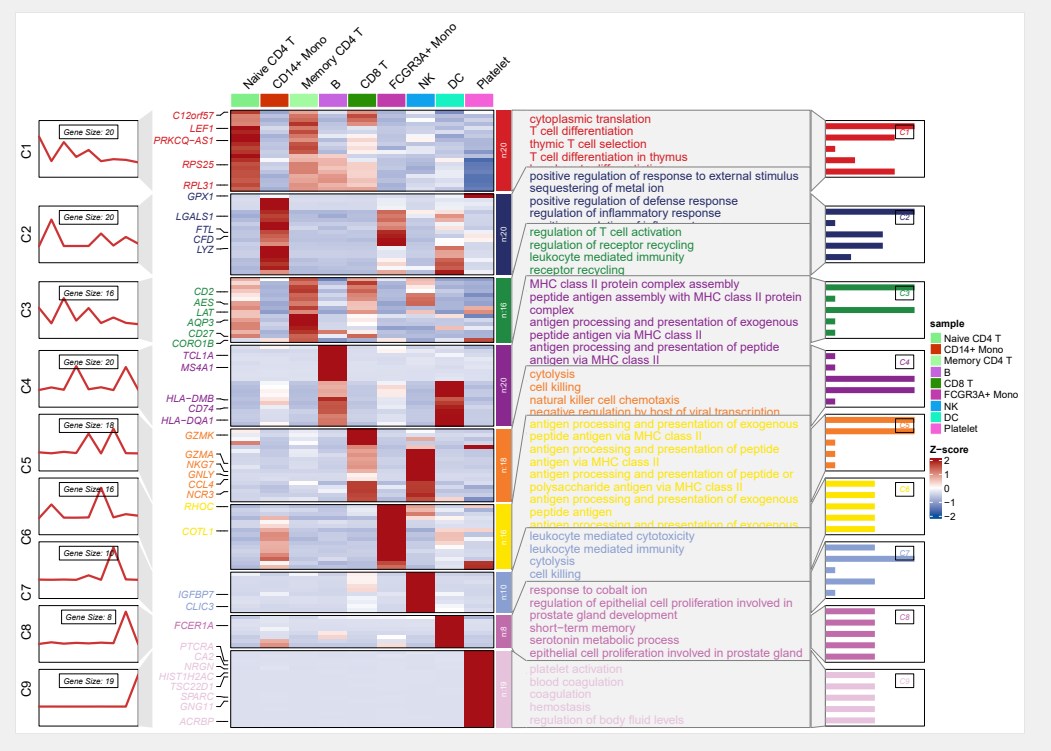
小果今天对ClusterGVis包的分享就到这里,该包对于转录组数据的可视化效果非常不错,可以用一张热图来展示基因的表达模式,聚类和功能注释,功能注释包括了常见的GO和KEGG,用起来超级方便,值得推荐,感兴趣的可以去github(https://github.com/junjunlab/ClusterGVis)深入学习,欢迎大家和小果一起讨论学习奥,今天的分享就到这里了,觉得不错的小伙伴可以点赞加关注奥,下期再见奥。