scCancer–你的肿瘤单细胞包转录组表分析师!
你知道有一个工具可以帮助你分析肿瘤单细胞的转录组吗?甚至这个工具可以直接帮你生成分析报表嘛?今天小果就带大家来认识这个操作简单,效率又极高的R工具包,学会后可以直接成为你的分析助理哦!现在大家就跟着小果一起来看看吧!
安装scCancer
是的没错,今天要带大家认识并学习的R工具包就是scCancer,首先让我们来一起学习下如何安装吧!小果在这里整理了两种安装方法,各位同鞋根据自己的情况自行选择下载哦!
- 通过Bioconductor下载
BiocManager::install(‘scCancer’)
 直接通过Bioconductor下载的同学要注意啦!这样下载对你的R版本是有要求的哦,如果版本不匹配会报以下错误信息,小果第一次在下载的时候就遇到了这样的问题。
直接通过Bioconductor下载的同学要注意啦!这样下载对你的R版本是有要求的哦,如果版本不匹配会报以下错误信息,小果第一次在下载的时候就遇到了这样的问题。
所以,小果有找到一种更不容易出错的下载方法:
- 通过github下载
install_github(‘linxihui/NNLM’)
install_github(“wguo-research/scCancer”)
这里小果温馨提示,通过gihub下载之前要通过github先下载好NNLM包哦,不然下载scCancer包的过程也会报错导致无法正常完成下载哦!
scCancer自动生成肿瘤细胞分析报表
- 下载单样本数据
在进入我们带的分析流程之前,小果为大家贴出以下数据连接作为我们分析的参考数据:http://lifeome.net/software/sccancer/KC-example.tar.gz
同学们赶紧跟着小果一起下载起来吧!~~~
下载好后,同学们要把这些数据解压在自己合适的工作目录下哦,并打开我们的数据文件夹,下面小果来带着大家一起认识一下这些数据的主要内容吧!
> list.files(“./sample”,recursive = T)
[1] “KC-example/filtered_feature_bc_matrix/barcodes.tsv.gz”
[2] “KC-example/filtered_feature_bc_matrix/features.tsv.gz”
[3] “KC-example/filtered_feature_bc_matrix/matrix.mtx.gz”
[4] “KC-example/raw_feature_bc_matrix/barcodes.tsv.gz”
[5] “KC-example/raw_feature_bc_matrix/features.tsv.gz”
[6] “KC-example/raw_feature_bc_matrix/matrix.mtx.gz”
首先,我们下载的数据均为单样本数据,其中raw_feature_bc_matrix代表过滤前的单细胞表达数据,filtered_feature_bc_matrix 代表过滤后的单细胞表达数据。
- 开始分析
现在我们完成了R包的安装和数据集的导入,小果要告诉你的是,其实我们已经完成了一大部分工作,因为scCancer可以自己完成肿瘤细胞的全分析流程,并为我们生成完整的分析报表在我们的结果目录中,我们接下来要做的就是配置所有的数据合保存路径即可,接下来我们一起来看一下如何操作吧!
dataPath <- “./data/sample” #设置输入的单细胞数据存放的路径
savePath <- “./results” #设置最后结果存放的目录
sampleName <- “sample” #细胞总名称
authorName <- “coucou” #报表作者名称,根据自己的修改哈
stat.results <- runScStatistics(
dataPath = dataPath,
savePath = savePath,
sampleName = sampleName,
authorName = authorName
)
以上的操作我们是基于runScStatistics 来计算QC过滤指标,scCancer会帮助我们生成完整的报表,整个分析流程大概序列要2~3分钟左右。
最后可以在结果文件中找到report-scStat.html查看所有分析结果,是不是特别简单高效!
在最后的报表中,我们可以看到对肿瘤细胞中的肿瘤微环境分析、恶性细胞评估、细胞周期评估、干细胞特征评估、基因集信息得分评估、表达程序识别、细胞间相互作用分析等信息,同学们只要从中提取对自己的研究有用的信息即可!
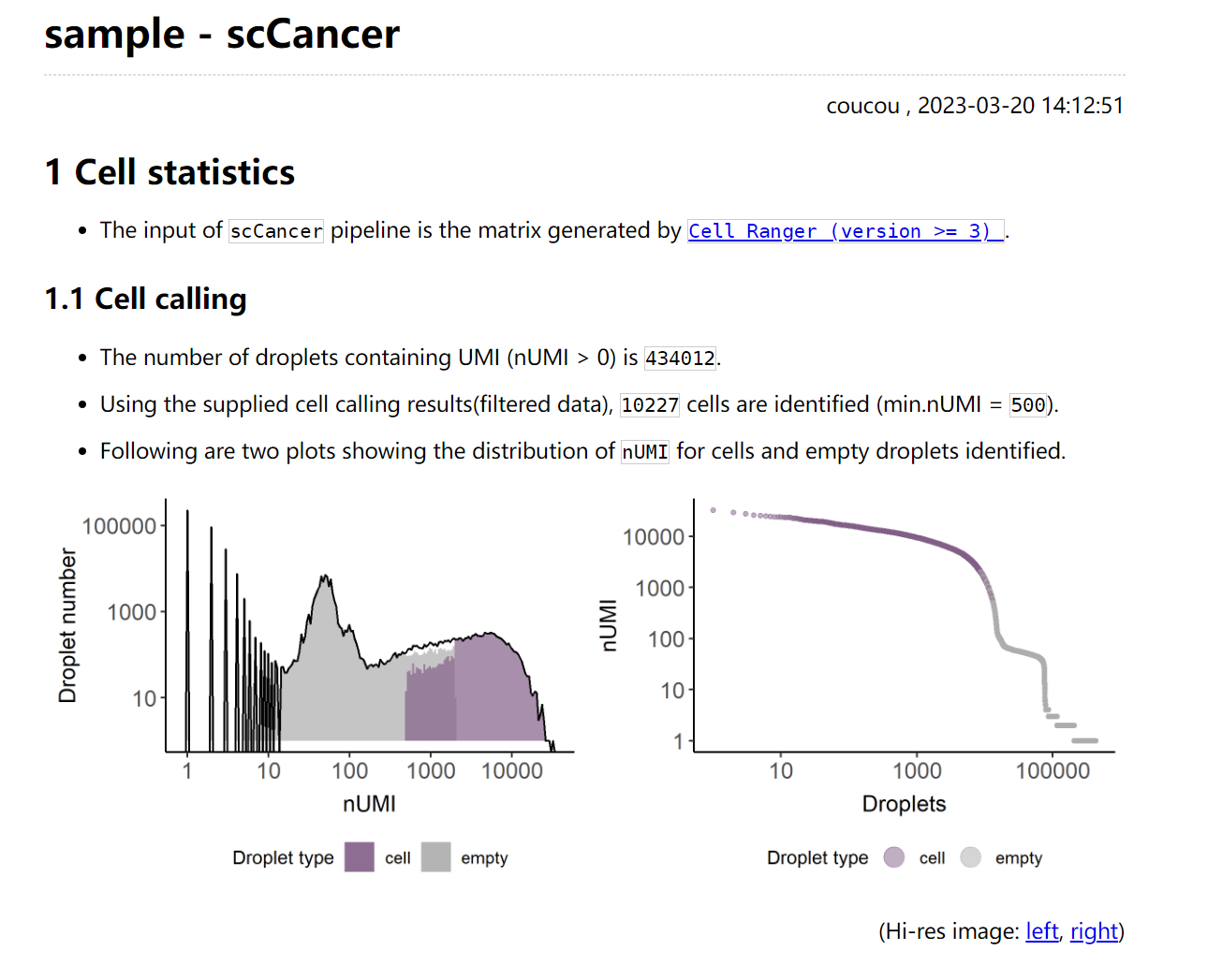
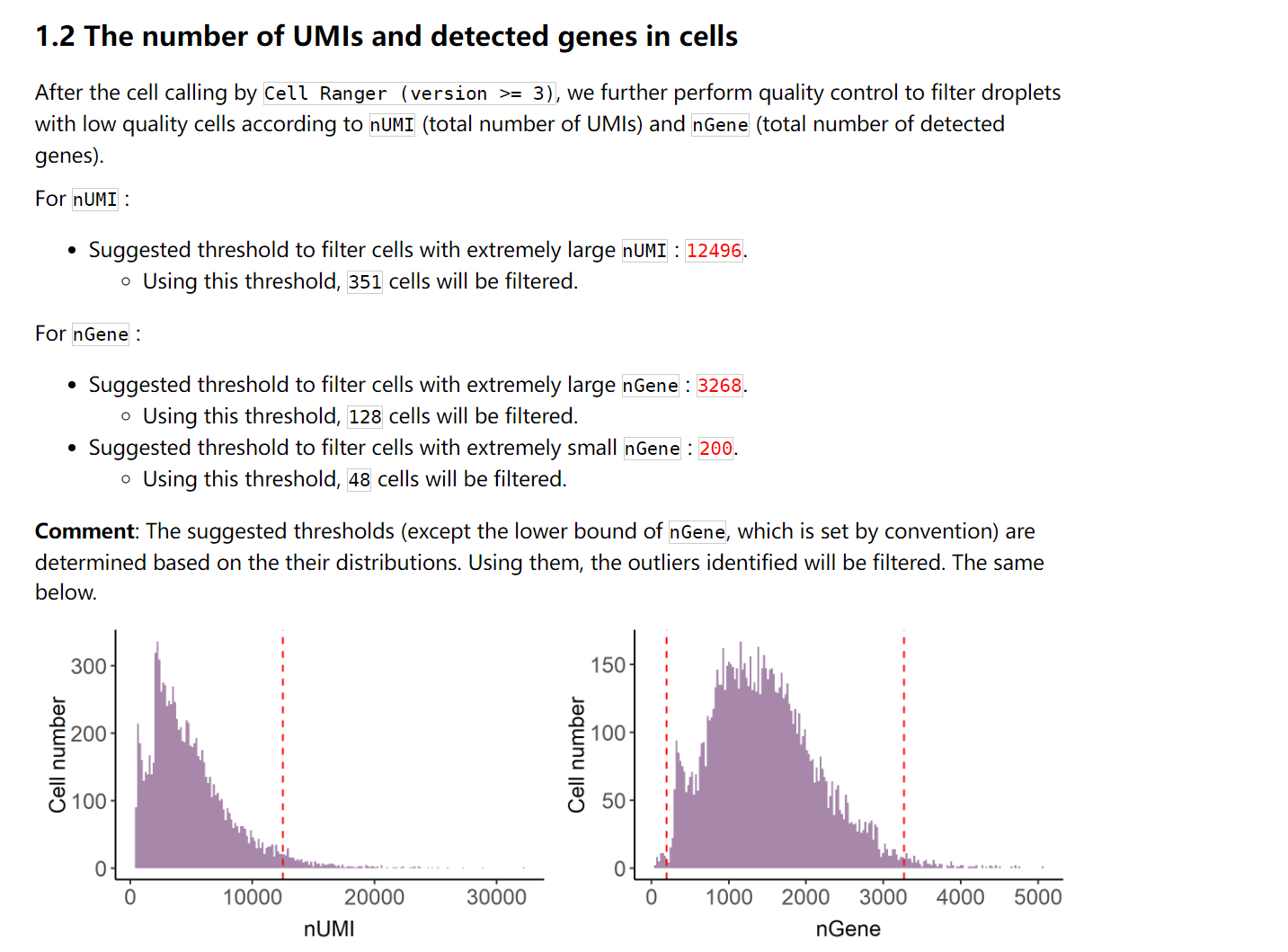
部分报表展示:
好啦,今天的小工具你学会了嘛?
更多生信工具的学习敬请继续关注小果哦!!