热图-生成你的单细胞基因表达“调色板”
当我们生成单细胞的数据矩阵并了解了细胞中基因表达的基本情况后,我们要如何整体观察细胞间基因表达量的相关性呢?大家一起来和小果看一下吧!
- 数据准备
library(scRNAseq)
data(fluidigm)
fluidigm<-ReprocessedFluidigmData()
ct <- floor(assays(fluidigm)$rsem_counts) #生成数据矩阵
ct[1:4,1:4]
sample_ann <- as.data.frame(colData(fluidigm))
DT::datatable(sample_ann)
View(ct)
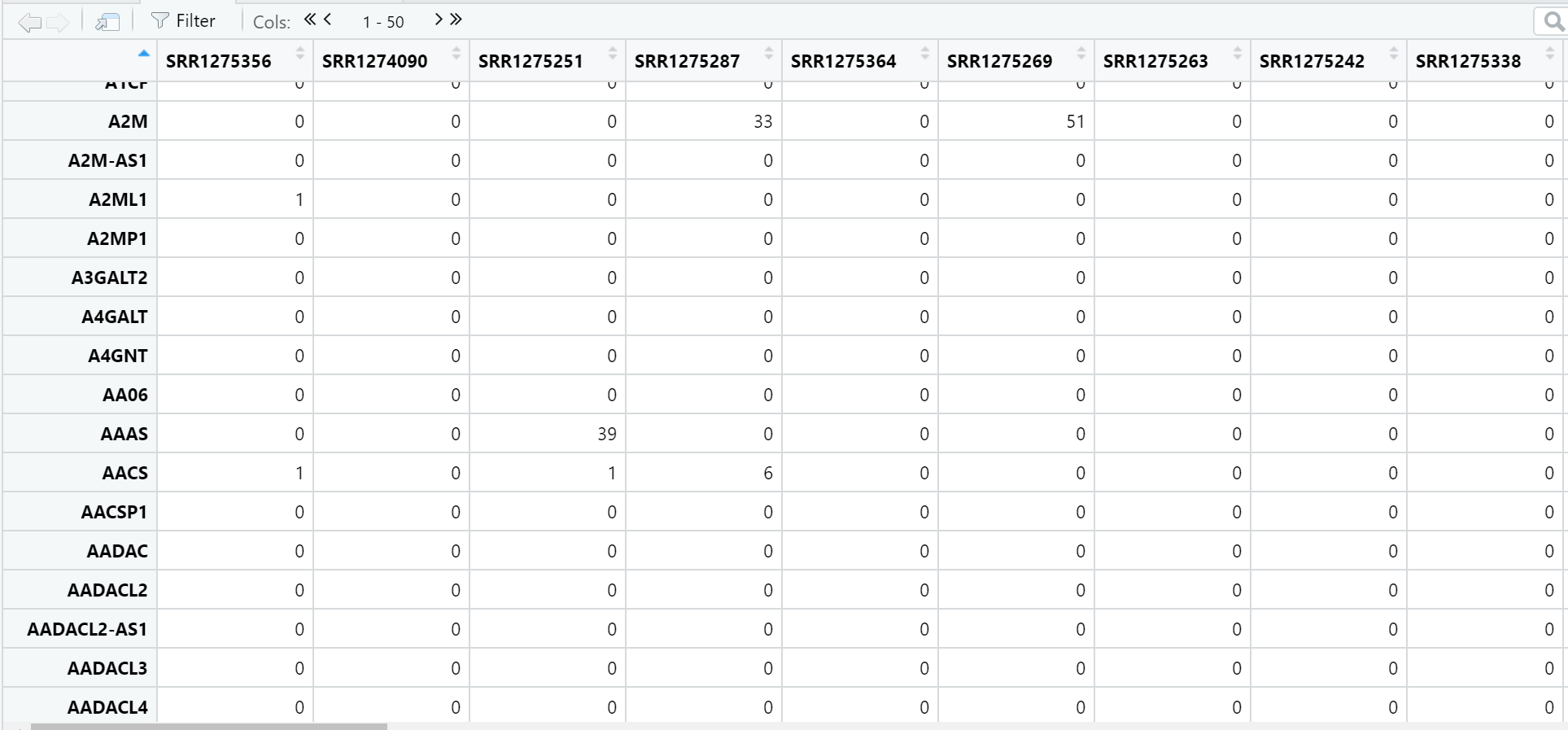 首先,我们先来一起看一下生成的数据矩阵:
首先,我们先来一起看一下生成的数据矩阵:
- 数据过滤
在查看基因在细胞中表达的相关性之前,我们先对数据做一个简单的筛选,在这里我们筛选出所有表达细胞数大于0的基因,和小果一起来看一下具体操作吧!
chose_gene= apply(counts,1,function(x) sum(x>0) )>0
table(chose_gene)
counts <- counts[chose_gene,]
table((apply(counts,1,function(x) sum(x>0)>0 )))
- 绘制热图
没错,现在我们来到了今天的重头戏,那就是通过热图来观察基因在细胞中表达的相关性和差异性,那么什么是热图呢?在绘制之前小果先给大家做一个简单的介绍:
-
- 热图的概念:
热图(Heatmap)是通过将数据矩阵中的各个表达值按照一定的规律映射为不同的颜色,最终呈现给我们一个像“调色盘”一样的图表来展示数据的变化规律,可以直接美观地向我们表达基因的全局表达状况哦。
-
- 绘制热图
要绘制本次实验中对应数据矩阵的热图,只需要通过R语言中的 pheatmap()函数绘制方法即可,让我们一起来看一下具体的代码操作吧!
#细胞之间所有基因表达量的相关性
data <- log2(edgeR::cpm(counts) +1)
data[1:4,1:4]
data_back <- data
experSet <- data_back
colnames(experSet)
#pheatmap 绘制热图的函数
pheatmap::pheatmap(cor(experSet))
让我们一起来看看最终呈现出的热图是什么样吧!
-
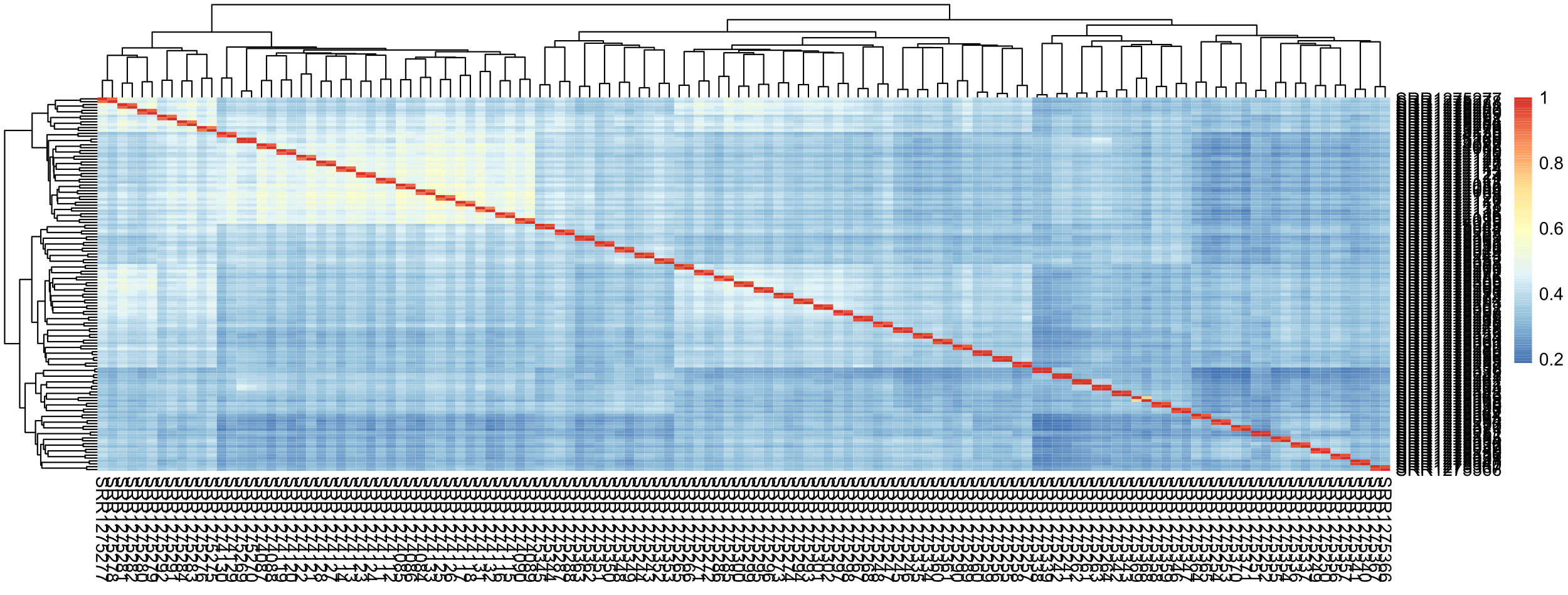 怎么分析热图?
怎么分析热图?
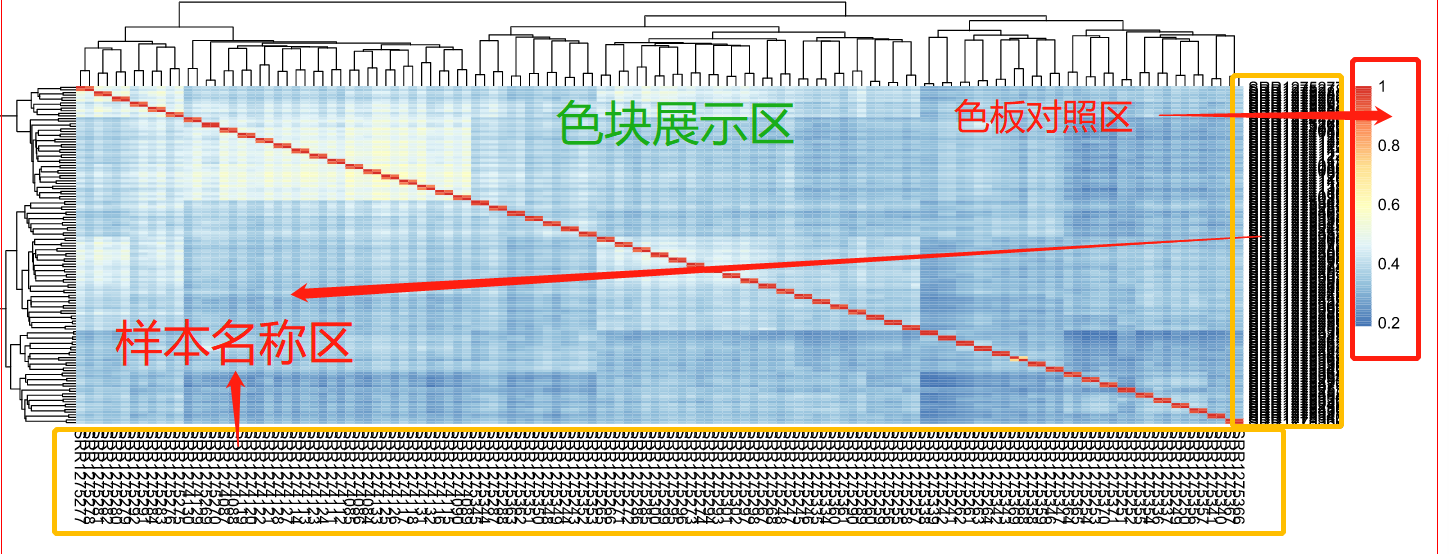 当我们成功绘制出细胞表达的热图后,如何快速地从中获取有效的信息呢?不要急,小果告诉你!
当我们成功绘制出细胞表达的热图后,如何快速地从中获取有效的信息呢?不要急,小果告诉你!
1.样本名称区:分为右侧和下方两部分,分别细胞名称和基因名称;
2.色块展示区:根据样本名称生成的颜色矩阵,每一个小矩形的色块 都代表一个表达数值;
3.色板对照区:色板中的每一个颜色都对应到每一个矩形中的颜色数值,在本图的色板对照区中,值越大越红,越小越蓝。
现在你学会热图的绘制以及分析方法了吗?同学们也可以自行下载一些数据,绘制属于你的专属“调色板”!大家也可以移步云生信画图专版进行更深入的学习哦!
小果云生信平台表达量热图绘制小工具www.biocloudservice.com/365/365.php
赶紧跟着小果一起学起来吧!