R语言带你探索单细胞表型信息
今天,小果要带大家继续探索单细胞测序相关R包的学习。之前为我们学习了scRNAseq包的安装和简单使用,那你知道如何使用它们并展示每个细胞基本的表型信息吗。那就和小果一起来看一下吧!
- scRNAseq包中有哪些数据?
首先,scRNAseq包中存放的是人类单细胞细胞,并将其分为四类细胞,这四类细胞分别是:
- neural progenitor cell (由pluripotent stem cells分化而成)
- GW16
- GW21
- GW21+3 (孕期细胞)
如果想要深入了解这些R包,同学们可以自行查阅资料了解,但如果是偏技术开发的同学也可以跳过这一部分的介绍哦,我们直接进入下面的正题。那就和小果一起来看一下吧!
- 数据载入
接下来,小果带大家一起通过scRNAseq包来导入数据。
library(scRNAseq)
data(fluid) #创建数据流名称
fluid<-ReprocessedFluidigmData() #从scRNAseq中导入数据
ct <- floor(assays(fluid)$rsem_counts)
ct[1:4,1:4]
sample <- as.data.frame(colData(fluid))
DT::datatable(sample)
 现在让我们一起看一下导入后的数据矩阵吧!
现在让我们一起看一下导入后的数据矩阵吧!
- 数据的表型信息
在上面的描述中提到,scRNAseq包中将人类单细胞分为四大类细胞,本次实验的数据集来自130个数据库,其中每个细胞的测序深度均不同,那么我们怎么能直观的看到每个细胞的具体统计指标呢?接下来小果用ggplot中的箱线图来给大家展示一下!
library(ggplot2)
box <- lapply(colnames(sample_ann[,1:19]), function(i){
dat <- sample_ann[,i,drop=F]
dat$sample=rownames(dat)
##绘制boxplot
ggplot(dat,aes(‘all cells’,get(i)))+
geom_boxplot()+
xlab(NULL)+ylab(i)
})
plot_grid(plotlist=box,ncol=5)
注意啦!最后可视化的plot_grid()函数来自cowplot包哦,同学们需要提前下载并导入哦,不然最后一定会报错!
现在让我们一起看看结果吧: 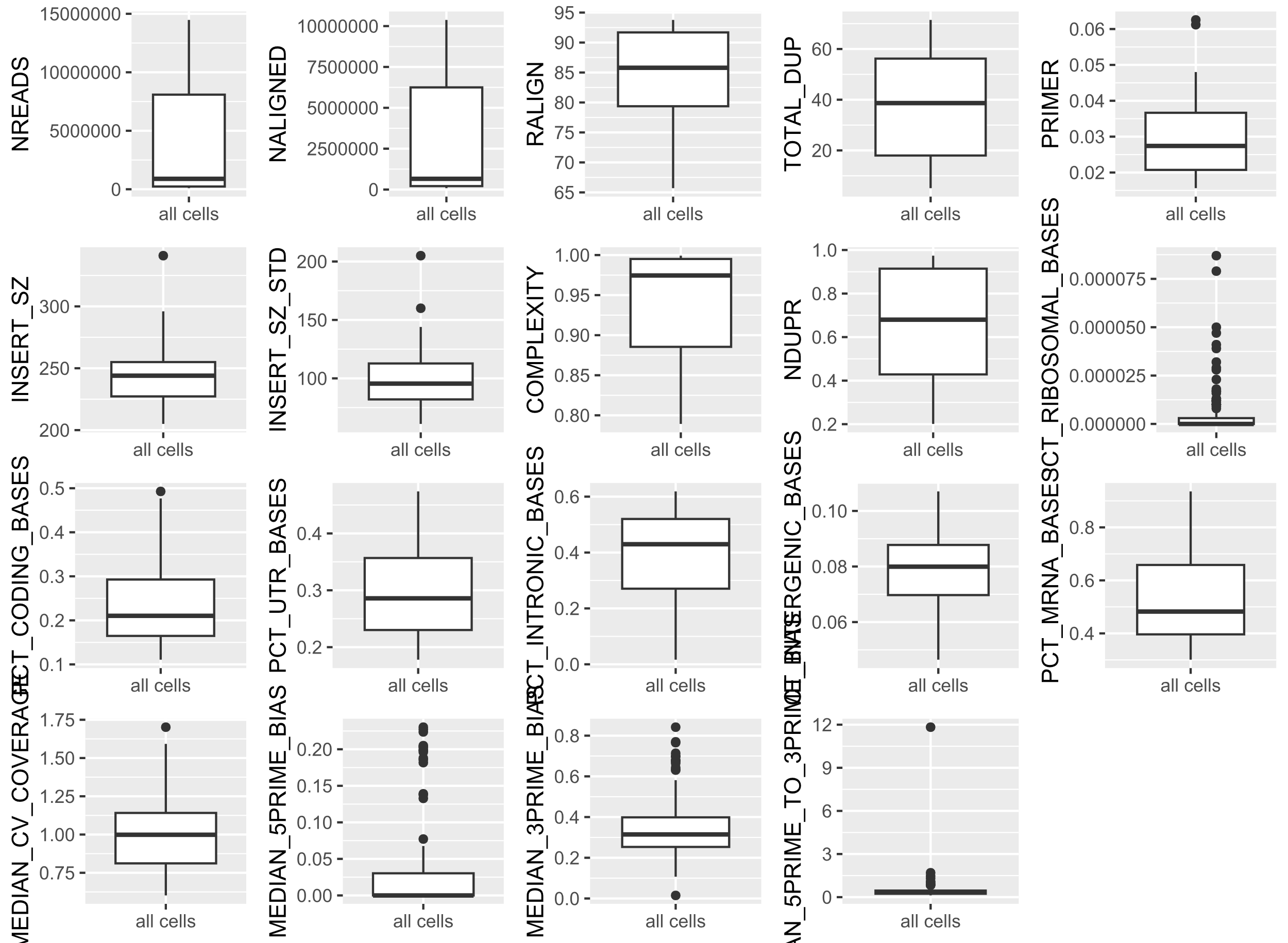
-
- 需要注意的小问题:
 小果在一开始进行可视化的时候,系统报错:
小果在一开始进行可视化的时候,系统报错:
因为想到同学们也可能和小果犯同样的错误,所以小果在这里先给大家做个预防!
 其实这不是什么很大的问题,如果经常用R工具包画图,就可能会报这样的错误哦,所以只要为我们把后台的图片缓存进行清理即可解决!
其实这不是什么很大的问题,如果经常用R工具包画图,就可能会报这样的错误哦,所以只要为我们把后台的图片缓存进行清理即可解决!