scran:细胞表达大数据的智能处理工具,助你成为表达分析专家!
在今天的学习之前,小果要和大家一起来回顾一下目前主流的scRNA-seq分析步骤,大家对单细胞分析的步骤是否进行过一些总结呢?没有也没关系,接下来就和小果一起来回顾一下吧!
- 通过主流数据库/第三方工具等方法下载分析所需要的测序数据;
- 预处理测序数据,通过预处理生成表达计数矩阵(行为基因/转录本,列为细胞),同时创建单细胞测试对象;
- 数据质控;
- 数据标准化;
- 数据筛选并分析;
- 数据降维和降噪;
- (看情况操作)整合多批次数据。
通过小果和大家的回顾,你想起来了这些流程了嘛?回顾过后,进入今天的正题,我们今天要一起学习的就是对scRNA-seq数据进行标准化处理的一个常用工具包:scran。接下来就让我们一起来看看scran是如何对数据进行标准化处理的吧!
scran的安装
BiocManager::install(‘scran’)
scran标准化数据与差异建模
- 数据准备
小果在这里使用由SingleCellExperiment包提供的人类胰腺数据集作为今天带的测试数据,这需要我们导入scRNAseq包,还没下载小伙伴赶紧下载起来哦!
library(scRNAseq)data <- GrunPancreasData()data()View(data)
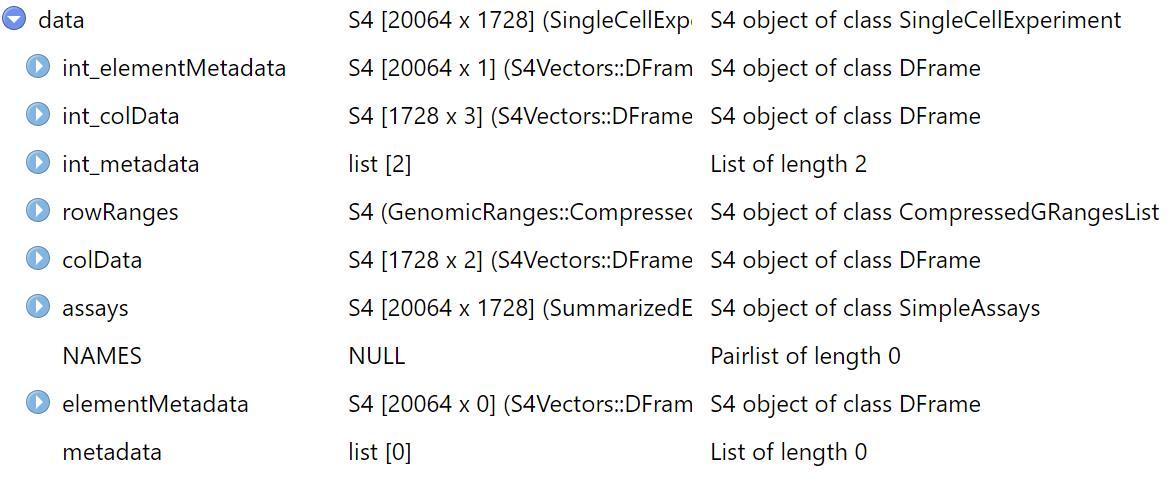 导入后,和小果一起来看下导入的数据结构吧!
导入后,和小果一起来看下导入的数据结构吧!
- 数据质控
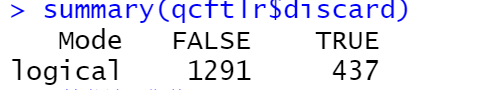
在标准化之前,我们首先对数据进行一个简单的略微的质控哦,我们的质控将过滤掉spike-in超高以及counts太低的细胞哦!
#简单的质控取除counts国第或者spike-in过高的细胞
library(scuttle)
qcdata <- perCellQCMetrics(data)
qcftlr <- quickPerCellQC(qcdata, percent_subsets=”altexps_ERCC_percent”)
data <- data[,!qcftlr$discard]
summary(qcftlr$discard)
- 数据标准化
在我们进行数据标准化处理之前,小果给大家简单的科普以下scran进行数据标准化处理的整体思路吧!
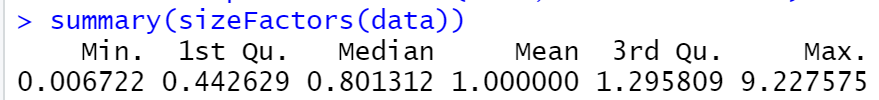
scran中的标准化会使用到computeSumFactors()函数,该函数的思路使用了“反卷积”的方法。针对单细胞测序的dropout和0计数现象,scran通过合并 (pool) 总计数类似的细胞,通过它们的计数总和来估算一个size factor,然后将其进一步分解,最后用到每个细胞表达谱的标准化中。
接下来就和小果一起来看一下具体的代码如何操作吧!
 #数据标准化
#数据标准化
library(scran)
clutrs <- quickCluster(data)
data <- computeSumFactors(data,clusters=clutrs)
summary(sizeFactors(data))
- 差异建模
基于上面数据的标准化处理,我们现在可以对数据进行差异化建模并可视化处理啦,还是和小果一起来看下如何操作吧!~
接下来就和小果一起来看一下具体的代码如何操作吧!
#数据标准化
library(scran)
clutrs <- quickCluster(data)
data <- computeSumFactors(data,clusters=clutrs)
summary(sizeFactors(data))
![]()
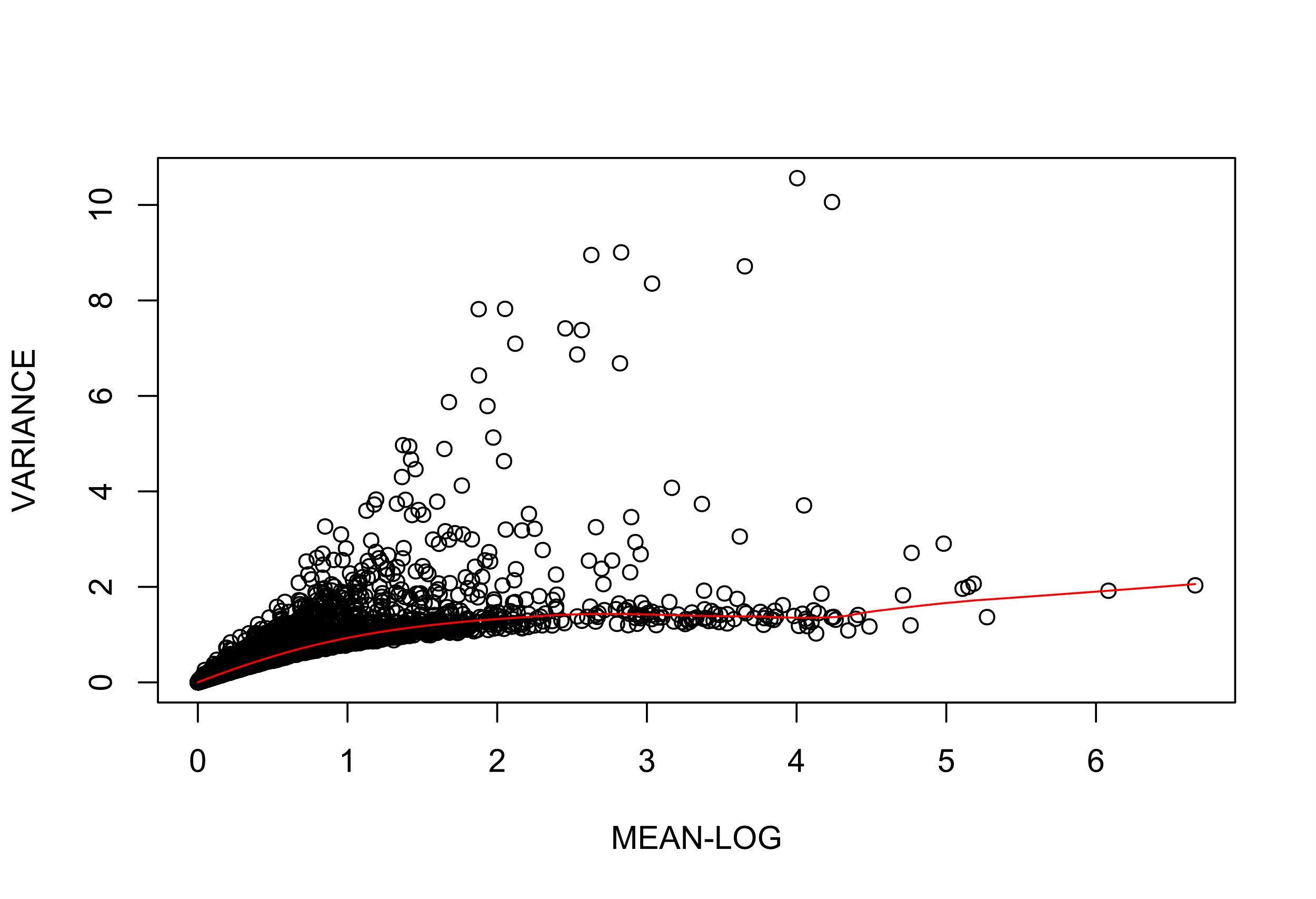
现在我们就得到了数据的差异模型表达图,那么如何分析这个图的统计结果呢?小果来给大家简单地做个解释吧!
首先,我们统计得到了每个细胞表达地平均值,为了方便计算统计,我们将他们取对数处理,y轴指的就是细胞之间的差异值,值越大,差异性越强。从图中可以看到,有少量的细胞分散在上方,但是大部分细胞的差异值基本都保持在0~5之间,整体的差异变化趋势呈平缓状态。怎么样,你看懂这个结果分析图了嘛?
好啦,今天的可视化工具你学会了嘛!?更多生信小工具的学习就请继续关注小果吧!下期我们不见不散!~~