我们生信人在做分析的时候,如果遇到的是单一类型的数据,如甲基化数据,转录组学数据、代谢组学数据等,我们可能是手拿把掐,信手捏来。如果我们遇到的是多种组学数据呢,我们可能还是每一类数据都给他单一分析一遍,哼哧哼哧分析一顿,每个类型都按照常规流程分析出来,当然可以,把结果一罗列,我们的分析内容也会显得很丰富。但是,大家有没有想过,这样单一的分析会忽略不同特征之间的关系,很有可能因此忽略关键的信息!难道我们就不能多组学数据共同分析吗?mixOmics帮我们解决了这么一大问题!它可以让我们将不同的组学数据进行预处理后共同分析,并将问题主要放在特征选择上,其在多组学领域知名度也是很高,文章引用量也很高,下图是该R包的文章:
 文中指出,研究团队推出的mixOmics,是一个专门用于生物数据集多变量分析的R包,特别关注数据探索、降维和可视化。通过采用系统生物学方法,该工具包提供了多种方法,可以同时统计整合多个数据集,以探索异构“组学”数据集之间的关系。简而言之,mixOmics主要有三种分析模式:
(1)在面对单类组学数据时,可以使用PCA,sPCA进行降维以及特征筛选,而面对有监督的数据类型时,可以转而使用PLS-DA,sPLS-DA在考虑样本分类信息的情况下将样本投射到低维空间中并进行分析。
(2)而面对多种不同类型组学数据时,可以使用N-集成(DIABLO)将不同数据联合起来,例如基因组,mRNA数据,蛋白组学以及代谢组学的联合分析。
(3)面对多组相同类型组学数据时,可以使用P-集成(MINT)方法,如对不同的蛋白组学数据进行联合分析。
今天小果会给大家展示该项目第一种分析模式的示例数据及代码,也是我们最常用的一种情况。特别提示,本次操作对电脑配置有一定需求,建议大家使用服务器运行,如果没有自己的服务器欢迎联系我们来租赁服务器哦~
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~
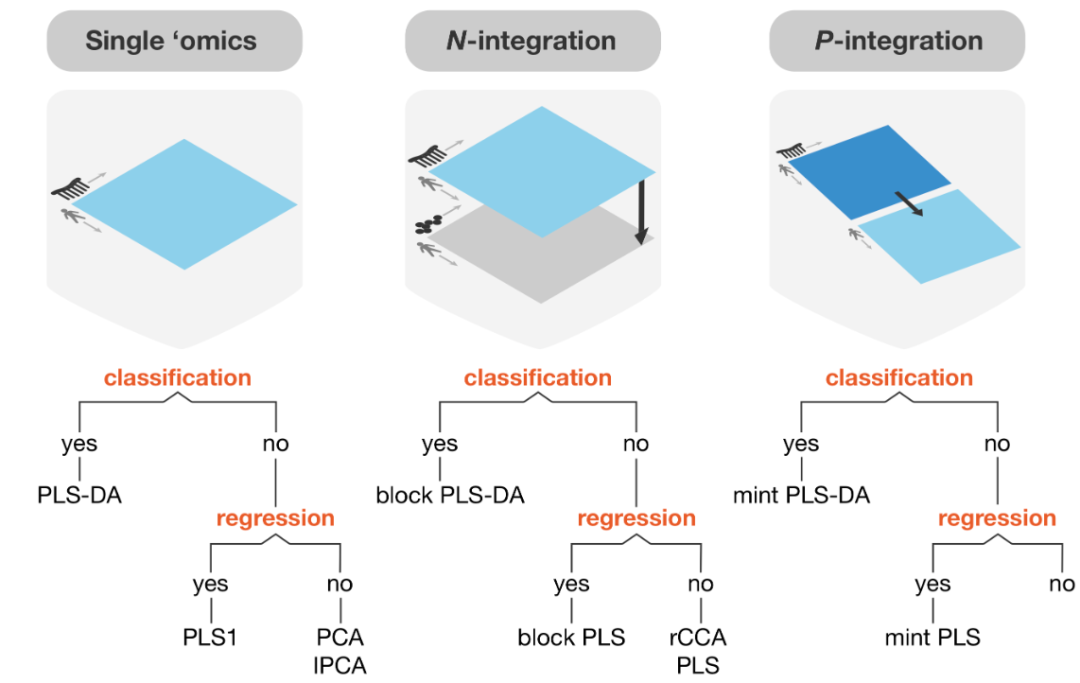
正式讲解流程之前,大家先看一下三种分析的示意图,该示意图很直观地展示了三种数据类型,方便大家进行理解,如下图:
文中指出,研究团队推出的mixOmics,是一个专门用于生物数据集多变量分析的R包,特别关注数据探索、降维和可视化。通过采用系统生物学方法,该工具包提供了多种方法,可以同时统计整合多个数据集,以探索异构“组学”数据集之间的关系。简而言之,mixOmics主要有三种分析模式:
(1)在面对单类组学数据时,可以使用PCA,sPCA进行降维以及特征筛选,而面对有监督的数据类型时,可以转而使用PLS-DA,sPLS-DA在考虑样本分类信息的情况下将样本投射到低维空间中并进行分析。
(2)而面对多种不同类型组学数据时,可以使用N-集成(DIABLO)将不同数据联合起来,例如基因组,mRNA数据,蛋白组学以及代谢组学的联合分析。
(3)面对多组相同类型组学数据时,可以使用P-集成(MINT)方法,如对不同的蛋白组学数据进行联合分析。
今天小果会给大家展示该项目第一种分析模式的示例数据及代码,也是我们最常用的一种情况。特别提示,本次操作对电脑配置有一定需求,建议大家使用服务器运行,如果没有自己的服务器欢迎联系我们来租赁服务器哦~
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~
正式讲解流程之前,大家先看一下三种分析的示意图,该示意图很直观地展示了三种数据类型,方便大家进行理解,如下图:
可以看到,左边是单一组学数据,根据是否有分类标签对应着不同的分析策略,中间是不同组学数据联合,进行预处理后同样根据标签有无来进行对应的分析,右边是相同组学数据的联合分析。我们可以发现,该工具包基本包含了我们处理组学数据的所有情况,那么接下来,我将给大家展示一下该工具包的简单操作,以及单一组学数据的示例分析。其他两种模式大家可以登陆官方文档自行查看哦~http://mixomics.org/
可以探索和集成不同类型的生物数据。该R包可以处理连续规模测量的分子特征(例如微阵列、基于质谱的蛋白质组学和代谢组学)或基于测序的计数数据(RNA-seq、16S、鸟枪法宏基因组学),这些数据在预处理和标准化处理后成为“连续”数据。
l归一化。该包不进行归一化处理,因为它是特定于平台的,并且涵盖的数据种类太多。在分析之前,该包假设数据集已使用适当的标准化方法进行标准化,并在适用时进行了预处理。
l预过滤。虽然该R包可以处理大型数据集(数万个预测变量),但我们建议将数据预过滤到每个数据集少于 10K 预测变量,例如使用中值绝对偏差进行RNA-seq数据,通过删除微生物组数据集中一贯的低计数或删除接近零的方差预测变量。该步骤旨在减少参数调整过程中的计算时间。
l数据格式。我们的方法使用矩阵分解技术。因此,数字数据矩阵或数据框在行中具有n个观测或样本,在列中具有p个预测变量,例如,基因、蛋白质、种下单元(OTUs)。
l协变量。在当前版本中,可能会混淆分析的协变量未包含在方法中。该R包建议事先使用适当的单变量或多变量方法校正这些协变量,以消除批次效应。
简而言之,就是需要我们提前将我们的数据进行处理,这和我们的常规流程处理数据其实是一样的。
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("mixOmics")
BiocManager::install("mixOmicsTeam/mixOmics")# 该mixOmics包应会直接导入以下包:igraph, rgl, ellipse, corpcor, RColorBrewer, plyr, parallel, dplyr, tidyr, reshape2, methods, matrixStats, rARPACK, gridExtra。对于Apple mac用户:如果无法安装导入的包rgl,则需要先安装XQuartz软件。
library(mixOmics)# 检查加载包时是否有错误,尤其是rgl库(见上文)
下面步骤给出的示例使用的数据已经是包的一部分。要上传您自己的数据,请首先检查您的工作目录是否已设置,然后通过使用RStudio 中的“文件”>“导入数据集”或通过以下命令行之一以.txt或.csv读取数据。
# from csv filedata <- read.csv("your_data.csv", row.names = 1, header = TRUE)# from txt filedata <- read.table("your_data.txt", header = TRUE)
data(nutrimouse) #R包自带示例数据X <- nutrimouse$gene
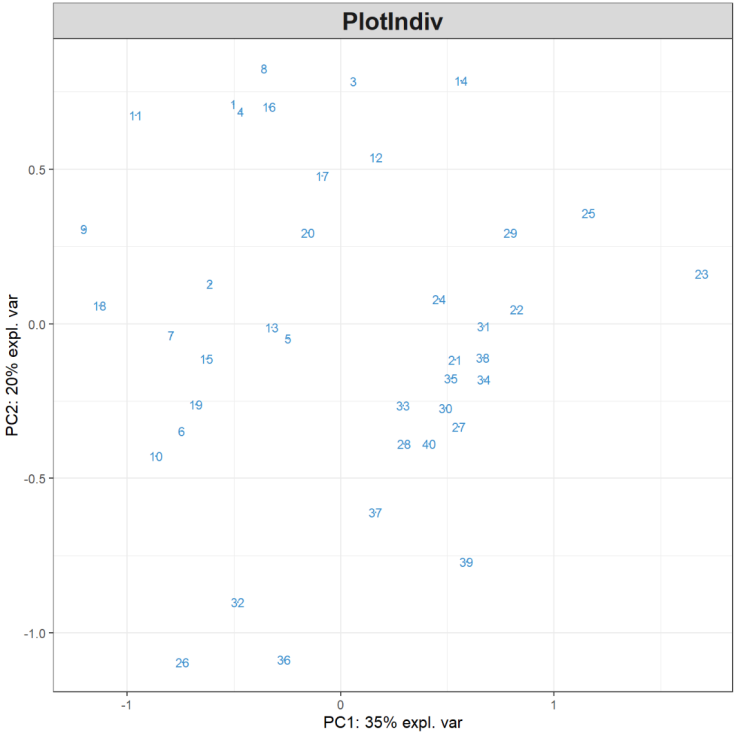
MyResult.pca <- pca(X) # 1 运行PCA方法plotIndiv(MyResult.pca) # 2 绘制样本图
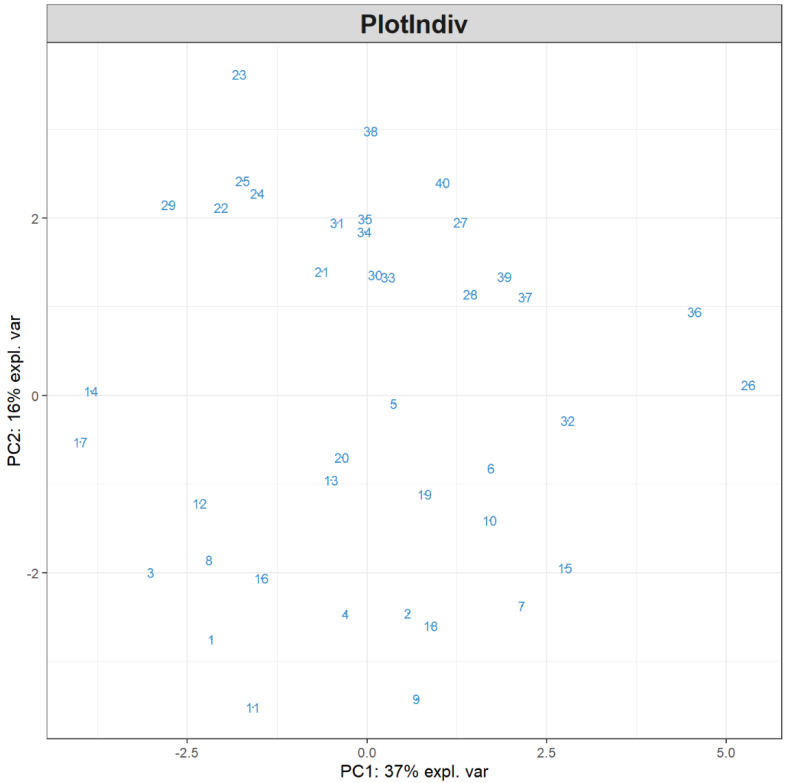
plotVar(MyResult.pca) # 3 绘制变量图
按照我们此处的示例,可以应用稀疏 PCA 来选择对 PCA 中的两个分量做出贡献的前 5 个变量。用户指定在每个组件上选择的变量数量,例如,这里在前两个组件上分别选择 5 个变量 (keepX=c(5,5)):
MyResult.spca <- spca(X, keepX=c(5,5)) # 1 运行PCA方法plotIndiv(MyResult.spca) # 2 绘制样本图
plotVar(MyResult.spca) # 3 绘制变量图
为了演示PCA和其变体,我们将分析multidrug包中提供的案例研究。这项药物基因组学研究调查了癌细胞系中的药物活性模式。这些细胞系来自美国国家癌症研究所(NCI)发育治疗计划建立的NCI-60人类肿瘤细胞系,用于筛选不同癌细胞系中化学化合物库的毒性。NCI-60包括源自结直肠癌(7个细胞系)、肾癌(8个细胞系)、卵巢癌(6个细胞系)、乳腺癌(8个细胞系)、前列腺癌(2个细胞系)、肺癌(9个细胞系)和中枢神经系统来源的癌症(6个细胞系),以及白血病(6个细胞系)和黑色素瘤(8个细胞系)。
$ABC.trans:包含通过定量实时 PCR (RT-PCR) 对每个细胞系测量的 48 种人类 ABC 转运蛋白的表达。
$compound:包含 1,429 种药物的活性,以 GI50 表示,即对测试细胞系的细胞生长产生 50% 抑制的药物浓度。
$comp.name:1,429 种化合物的名称。
$cell.line:有关细胞系名称 ( $Sample) 和细胞系类型 ( $Class) 的信息。
输入的数据是矩阵类型,其中行名为每个样本的名称,列名是变量的名称(例如基因),我们从数据$ABC.trans开始:
library(mixOmics)data(multidrug)X <- multidrug$ABC.transdim(X) # 检查数据的维度结果:## [1] 60 48
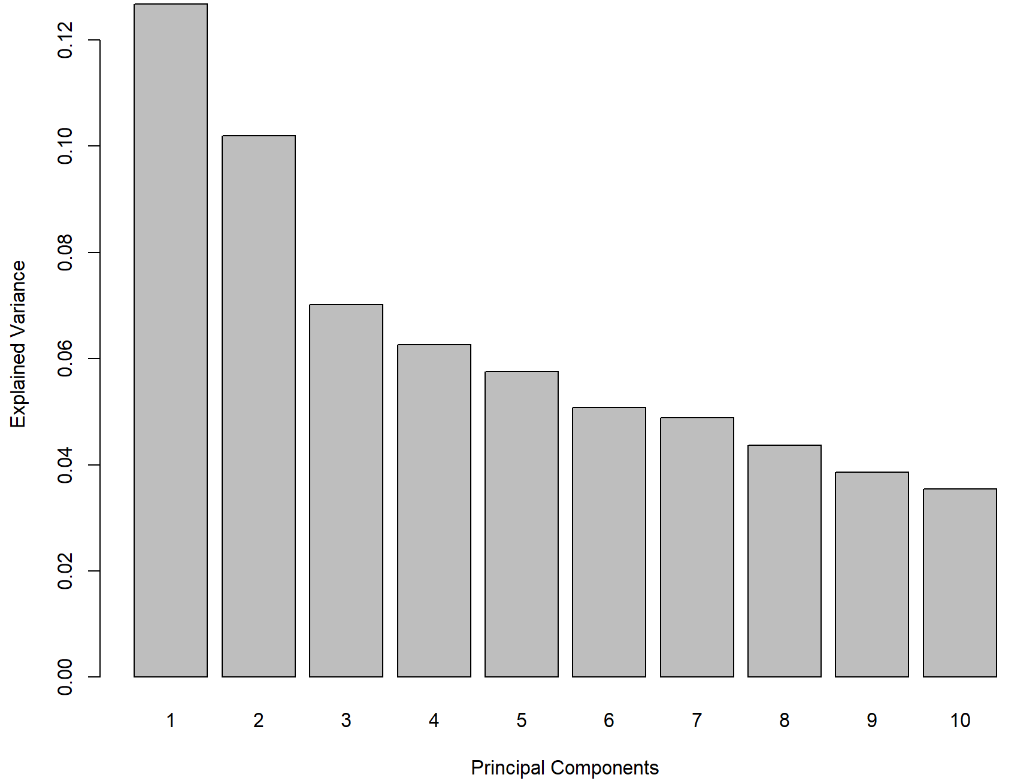
tune.pca()计算大量主成分的解释方差的累积比例(这里我们设置ncomp = 10)。输出解释方差相对于每个主成分数据方差总量的比例的细线图:
tune.pca.multi <- tune.pca(X, ncomp = 10, scale = TRUE)plot(tune.pca.multi)
图示为ABC 转运蛋白数据上每个主成分的解释方差量
tune.pca.multidrug$cum.var # 输出累计差异比例
我们根据上图的下降趋势,或者根据上一条给出代码计算的具体比例数值,确定前两个或者前三个主成分可以保留下来。
final.pca.multi <- pca(X, ncomp = 3, center = TRUE, scale = TRUE)
final.pca.multi$var.tot结果:## [1] 47.98305
通过对所有可能分量解释的方差求和,我们将获得相同数量的解释方差。每个分量的解释方差比例为:
final.pca.multi$prop_expl_var$X## PC1 PC2 PC3## 0.12677541 0.10194929 0.07011818
final.pca.multi$cum.var结果:## PC1 PC2 PC3## 0.1267754 0.2287247 0.2988429
为了计算分量,我们使用加载向量中指示的可变系数权重。因此,加载向量中系数的绝对值告诉我们每个变量对每个分量的定义的重要性。我们可以通过函数提取此信息,selectVar()该函数根据每个主成分的绝对负载权重值按降序排列最重要的变量。
#只根据第一主成分排序变量:head(selectVar(final.pca.multi, comp = 1)$value)结果:## value.var## ABCE1 0.3242162## ABCD3 0.2647565## ABCF3 0.2613074## ABCA8 -0.2609394## ABCB7 0.2493680## ABCF1 0.2424253
我们将样本投影到主成分所在的空间中,以可视化样本如何聚类并评估数据中的生物或技术变化。
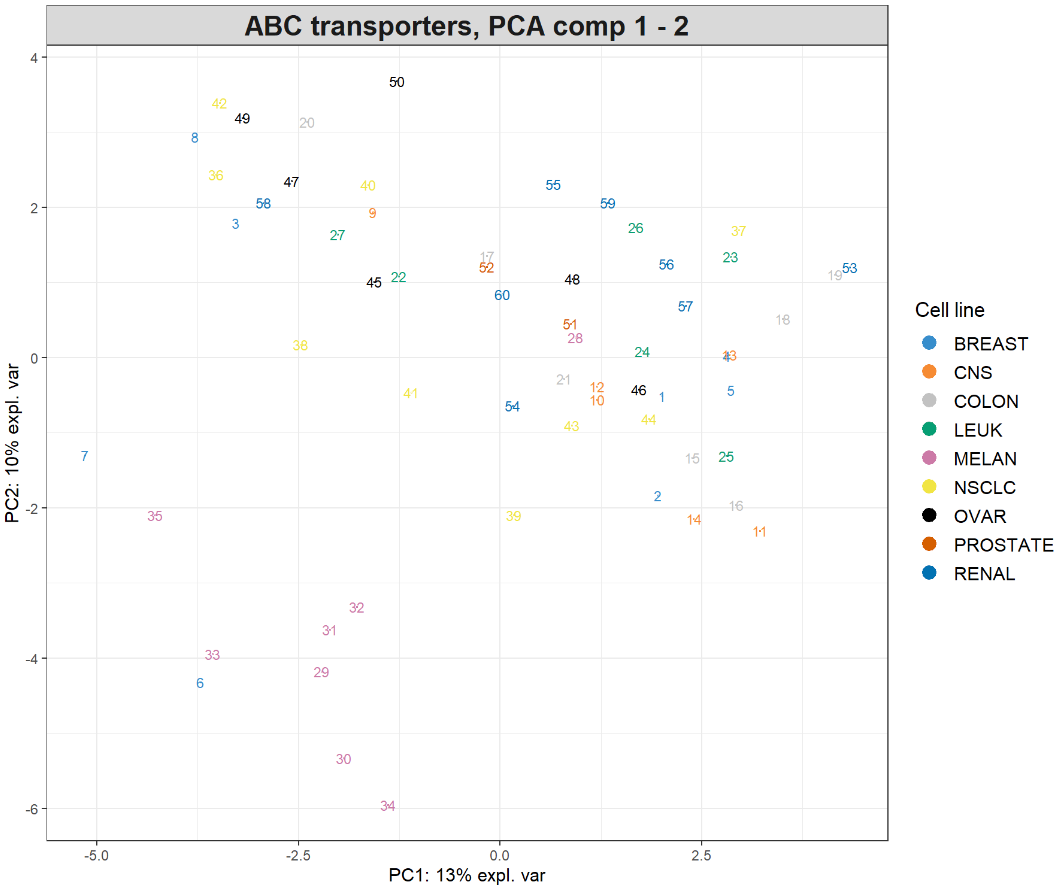
plotIndiv(final.pca.multi, comp = c(1, 2), # Specify components to plot ind.names = TRUE, # Show row names of samples group = multidrug$cell.line$Class, title = 'ABC transporters, PCA comp 1 - 2', legend = TRUE, legend.title = 'Cell line')
样本被投影到由前两个主成分跨越的空间中,并根据细胞系类型着色。数字表示数据的行名称。我们也可以进一步检查第三个主成分,只需修改comp = c(1, 3)即可,或者使用3D交互图,代码如下:
# 交互式3D绘图将加载rgl库.plotIndiv(final.pca.multi, style = '3d', group = multidrug$cell.line$Class, title = 'ABC transporters, PCA comp 1 - 3')
大家做过第三个主成分绘图后可发现,前两个分量解释的最大变异源可归因于黑色素瘤细胞系(MELAN),第三个分量只突出显示了单个异常样本。因此,对数据的解释主要集中在前两个主成分。
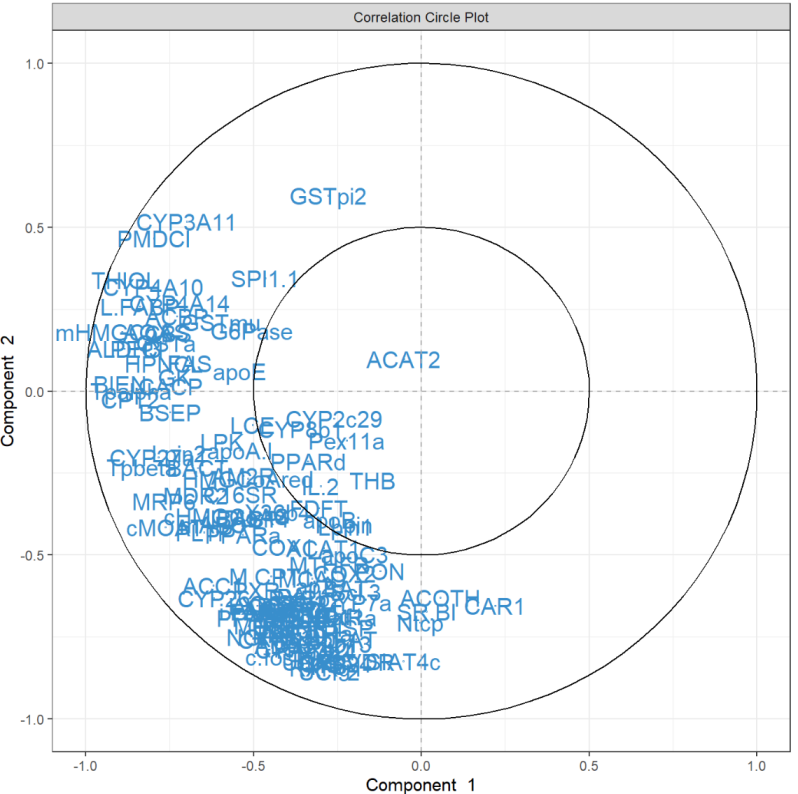
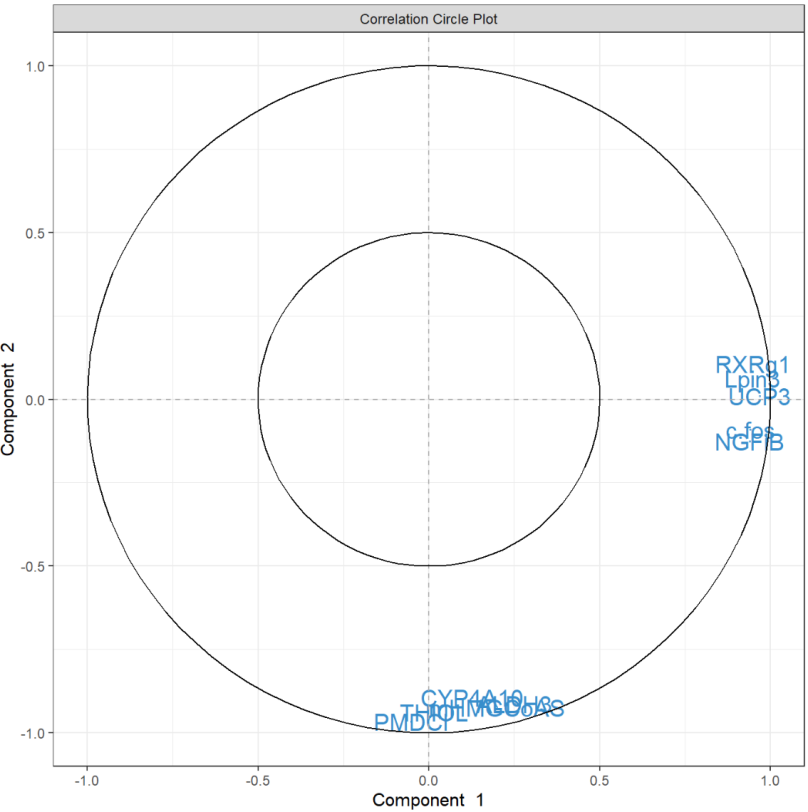
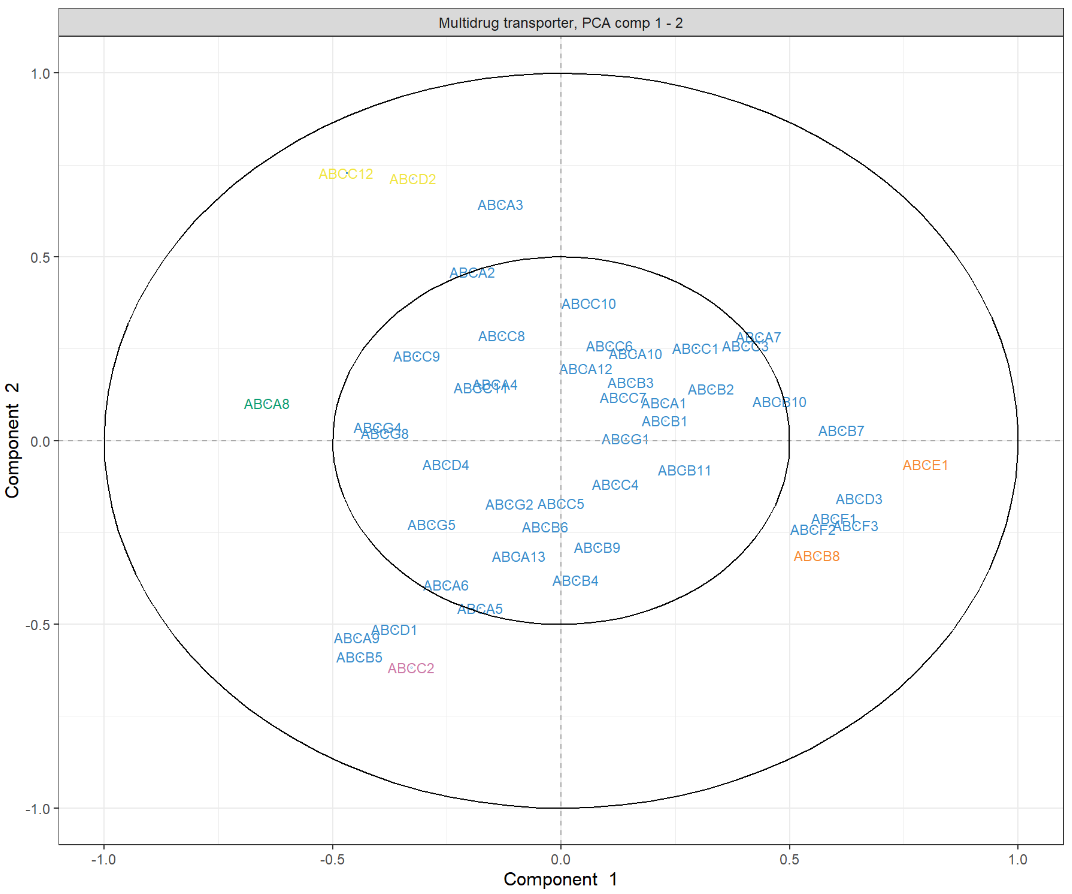
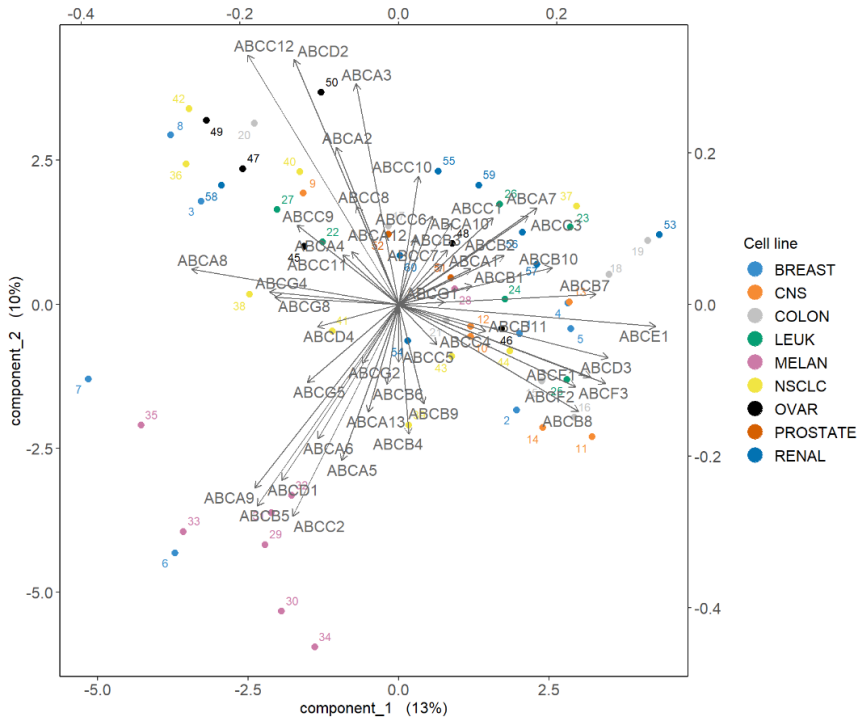
相关圆图使用该函数指示每个变量对每个分量的贡献plotVar(),以及变量之间的相关性(由变量“簇”表示)。
plotVar(final.pca.multi, comp = c(1, 2),var.names = TRUE, cex = 3, # To change the font size# cutoff = 0.5, # For further cutoff title = 'Multidrug transporter, PCA comp 1 - 2')
图中突出显示了一组对PC1有贡献的ABC转运蛋白:ABCE1,并且在某种程度上与ABCB8聚集的组对PC1有正向贡献,而ABCA8有负向贡献。我们还观察到一组对 PC1 和 PC2 都有贡献的转运蛋白:与ABCC2聚集的一组对 PC2 有正向贡献,对 PC1 有负向贡献,而ABCC12和ABCD2簇对 PC1 和 PC2 都有负向贡献。
双图允许我们同时显示样本和变量,以进一步了解它们的关系。样本显示为点,而变量显示在箭头的尖端。与相关圆图类似,数据必须居中并缩放以解释变量之间的相关性(作为变量箭头之间的余弦角)。
biplot(final.pca.multi, group = multidrug$cell.line$Class, legend.title = 'Cell line')
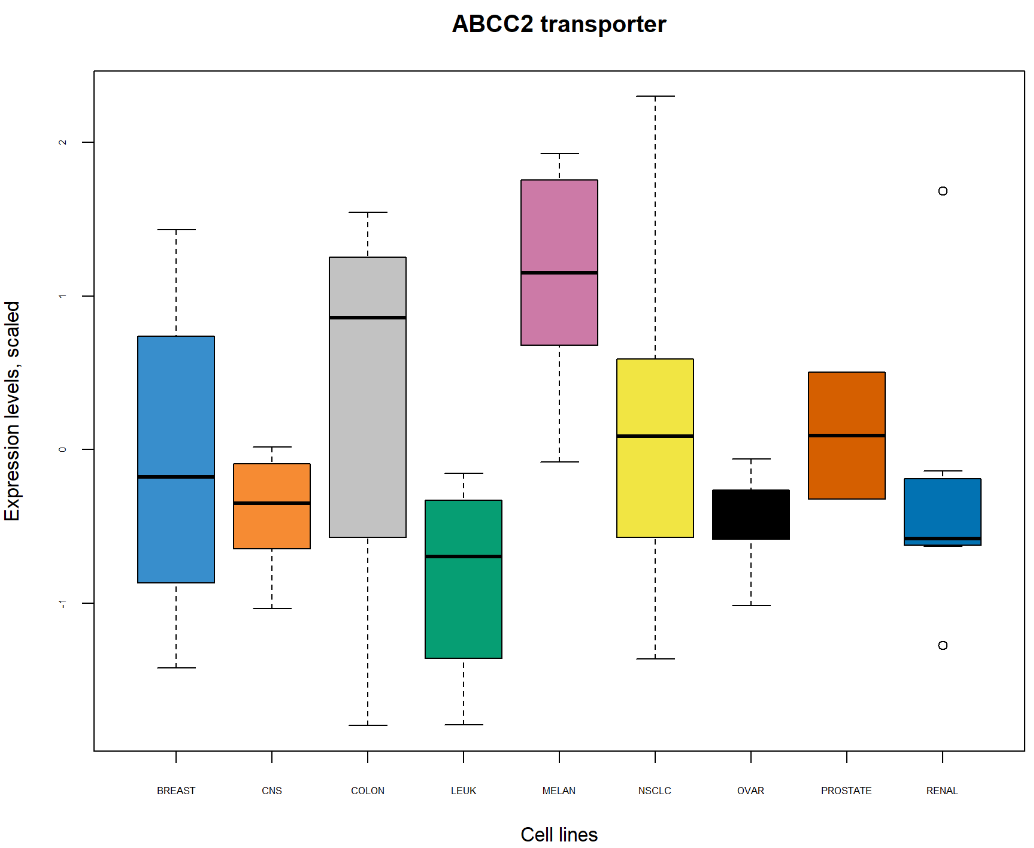
Biplot显示,黑色素瘤细胞系(MELAN)似乎以转运蛋白子集为特征,例如ABCC2周围的簇,如之前在样本图中突出显示的那样。进一步检查数据,例如箱线图,可以进一步阐明这些特定样本的转运蛋白表达水平。
ABCC2.scale <- scale(X[, 'ABCC2'], center = TRUE, scale = TRUE)boxplot(ABCC2.scale ~ multidrug$cell.line$Class, col = color.mixo(1:9), xlab = 'Cell lines', ylab = 'Expression levels, scaled', par(cex.axis = 0.5), # Font size main = 'ABCC2 transporter')
从PCA相关圆图和Biplot鉴定的转运蛋白ABCC2的箱线图显示与细胞系类型相关的ABCC2表达水平。
今天小果给大家展示的是mixOmics包的安装,快速使用以及PCA的示例分析。大家可以在自己的组学数据上进行尝试,今天只给大家展示了单一组学PCA的示例分析,大家如果对其他两种模式分析有兴趣的话,可以登陆该R包的官方文档或者原始文献再详细研究一下哦~
如果需要我们帮忙来复现该篇文献或者分析自己的数据都可以来找小果哦!今天小果的分享就到这里,如果小伙伴有其他数据的分析需求,可以尝试本公司新开发的生信分析小工具云品台,里面有成熟的代码,只要输入合适的数据就可以直接出想要的图呢,链接:http://www.biocloudservice.com/home.html。
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

 文中指出,研究团队推出的mixOmics,是一个专门用于生物数据集多变量分析的R包,特别关注数据探索、降维和可视化。通过采用系统生物学方法,该工具包提供了多种方法,可以同时统计整合多个数据集,以探索异构“组学”数据集之间的关系。简而言之,mixOmics主要有三种分析模式:
文中指出,研究团队推出的mixOmics,是一个专门用于生物数据集多变量分析的R包,特别关注数据探索、降维和可视化。通过采用系统生物学方法,该工具包提供了多种方法,可以同时统计整合多个数据集,以探索异构“组学”数据集之间的关系。简而言之,mixOmics主要有三种分析模式: