模式生物也能看功能,非人物种的富集分析

富集分析是我们科研分析中常见的一种分析方式,人的分析过程已经很清楚了,而且有在线工具可以意见富集,但一些非人物种,还需要借助一些代码来完成,富集分析中较常见的就是clusterProfiler,下面就让我们以小鼠为例,来看看富集分析是怎么做的吧。

代码如下
library(clusterProfiler) #加载程序包library(org.Mm.eg.db)dif_enrich<- read.table("T2DMvsCon.txt",header = T,row.names = 1,sep = "t")#读取表达文件gene <- rownames(dif_enrich)# 转为向量## 将gene symbol转换为Entrez IDgene.id <- bitr(gene, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Mm.eg.db")#org.Mm.eg.db包里记载了小鼠基因ID的对应关系egoBP<-enrichGO(gene = gene.id$ENTREZID, pAdjustMethod ="none",OrgDb = 'org.Mm.eg.db',ont = "BP", #GO富集有多种分类,其中最重要的就是BP,也就是生物学过程,还可以选择MF分子功能和CC细胞结构。如果全都要的话可以选择ALLpvalueCutoff = 0.05, #P小于0.05,即为显著富集,这里的设置就是只保留显著富集的结果,如果想把不显著的也都保存下来,这里可以选择把0.05改成1qvalueCutoff=1,minGSSize = 10,readable= TRUE)summary(egoBP) # 结果展示bp2 <- simplify(egoBP, cutoff=0.7, by="p.adjust", select_fun=min) #对富集到的GO项进行简化处理。write.table(summary(bp2), "GO.BP.txt", sep="t") #将富集结果写入文件pdf("GO.bp.pdf",height=8,width=8)barplot(bp2, showCategory=8) #条形图 showCategory这个参数是用来控制显示的GO项的数量的,默认是10,具体数量可根据需要来调整dev.off()pdf("GOdot.bp.pdf",height=8,width=8)dotplot(egoBP)#气泡图dev.off()
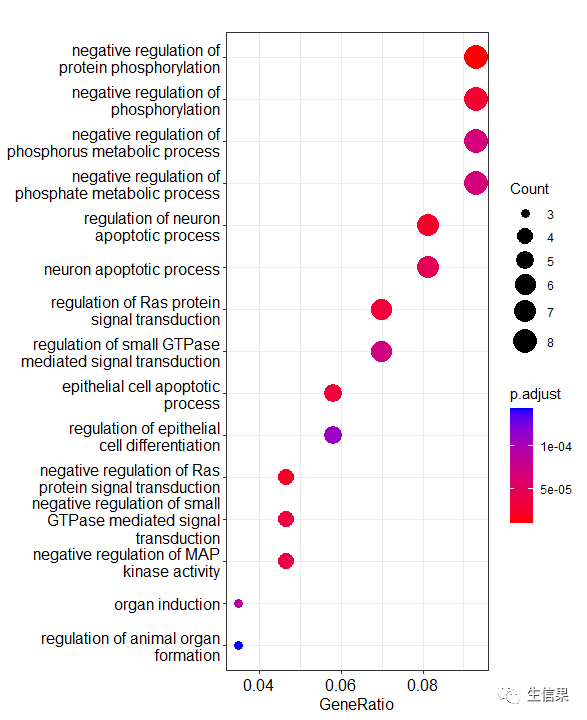
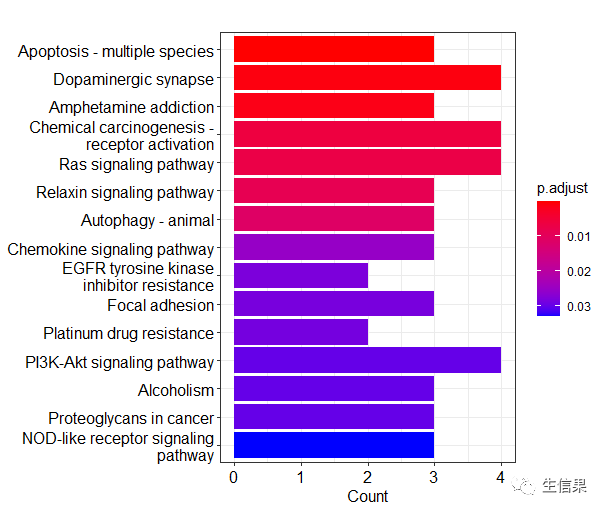
egoPathway<-enrichKEGG(gene = gene.id$ENTREZID, organism = "mmu", pvalueCutoff = 0.05,pAdjustMethod ="none",minGSSize =2,qvalueCutoff = 1) #KEGG富集,与GO富集类似,mmu代表小鼠summary(egoPathway) # 结果展示pdf("pathway.pdf")barplot(egoPathway, showCategory=8)dev.off()pdf("pathwaydot.pdf")dotplot(egoPathway)dev.off()
TIPS
公众号回复“111”获取文章相关文件
好了,这就是今天的主要内容了,代码还是比较简单的,小伙伴们有什么问题的话欢迎来和小云分享讨论哟。
推荐阅读
扫码关注我们

shengxinguoer
生信果
生信硬核知识解答
和小果一起学生信