掌握ggplot2,轻松绘制基因区间长度图!——R语言实战

相信大家都对R语言中的ggplot2包有所了解,那么你们知道怎么通过ggplot2绘制基因组的各染色体上的区间类型分布图嘛?今天小果就来给大家演示一下!
数据集准备
在绘制之前,我们首先要准备要导入的数据集,我们可以根据自己的情况将各染色体上进行“分区”,并汇总成一个table文件,我们一起来看看小果准备的数据文件吧!
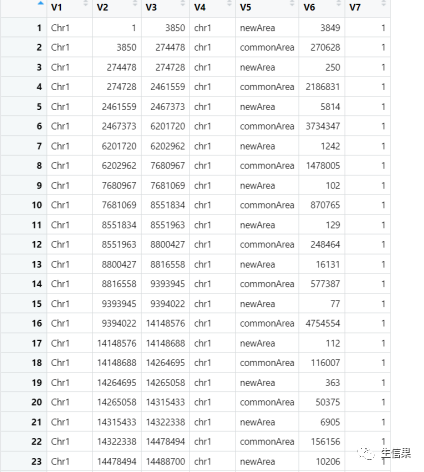
在这个数据集中,从左向右分别代表染色体编号、区域起点、区域终点、染色体名称、区域类型、区域长度、区域号。怎么样,你看懂了嘛?
导入数据集以及数据处理
首先,让我们看一下这段代码的作用。这段代码可以读取一个名为”newGeneArea-15.txt”的数据文件,并绘制出柱状图,其中每个柱子代表一段基因区域,横坐标表示基因所在的染色体编号,纵坐标表示基因区域的长度,不同颜色的柱子代表不同的基因分段类别,例如重要性高、中、低等。
下面,我将一步一步地为大家解释这段代码的实现过程。
1. 导入ggplot2包
library(ggplot2)library(ggplot2)
ggplot2是R语言中常用的可视化包,它提供了很多种图形类型和参数设置,可以用来绘制高质量的数据可视化图形哦。
2. 读取数据文件
a<-read.table(file = "newGeneArea-15.txt", header=F,quote="",sep="t",stringsAsFactors=F)3. 提取染色体号并加上”Chr”前缀
g<-sprintf("%s",unique(a[,4]))这行代码使用unique函数选出数据中唯一的染色体号,然后使用sprintf函数将它们转换为字符串,并在每个字符串前加上”Chr”前缀。这样做是为了后续用这些字符串来标记x轴。
4. 计算基因区域长度
a[,6]<-a[,3]-a[,2]这行代码将数据表a中的第6列设置为第3列减去第2列的差,进而得到每一个基因区域的长度。
5. 根据染色体号设置对应的横坐标上的位置列:
cssCopy codefor(i in 1:length(a[,1])){if(a[i,4] == "chr1"){a[i,7]=1}else if(a[i,4] == "chr2"){a[i,7]=2}else if(a[i,4] == "chr3"){a[i,7]=3}else if(a[i,4] == "chr4"){a[i,7]=4}else if(a[i,4] == "chr5"){a[i,7]=5}}
绘制图形
现在,我们已经完成了数据的预处理和整理,并且已经定义了我们要使用的图形元素和配色方案。现在,让我们用 ggplot2 包中的 ggplot 函数创建一个绘图对象,用于绘制我们的图形。我们将在此函数中指定数据框表a 作为数据源,同时指定在 x 和 y 轴上绘制的变量。
p <- ggplot(a, aes(x = a[,7], y = a[,6], fill = a[,5], group = a[,4])) + #指定变量 a[,7] 作为 x 轴变量,a[,6] 作为 y 轴变量geom_bar(stat = "identity") +theme_bw() + #为绘图对象添加一个基本的白色背景theme(panel.grid.major = element_blank(), #使用 element_blank 函数来隐藏 x 轴和 y 轴上的主要网格线legend.position = c(0.9, 0.9),legend.title = element_blank()) +scale_fill_manual(values = colors) + #函数设置条形填充颜色scale_x_continuous(breaks = c(1:5), labels = g) +labs(x = "", y = "") + #定义 x 轴的刻度值scale_y_continuous(labels = c(0, "10MB", "20MB", "30MB")) #定义 y 轴的刻度值
在上述的代码中,我们使用 geom_bar 函数绘制条形图。我们使用 “identity” 参数,以使 ggplot2 包知道我们已经使用了预处理的条形高度。我们还使用 theme 函数来进一步定制绘图对象的外观。
好啦,我们今天的绘图工作已经基本完成,我们一起来和小果看一下绘制好的图长什么样子吧!
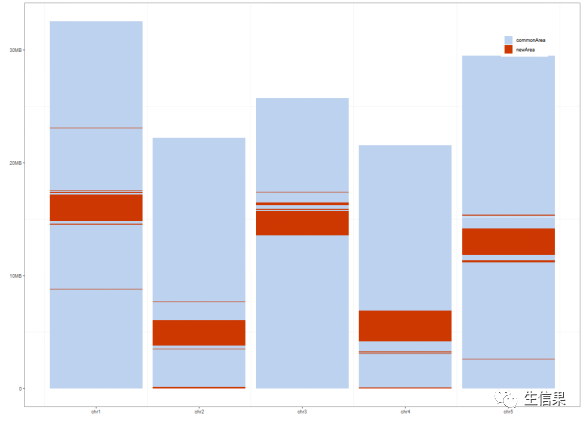
图片描述
在这个图中,主要向大家展示了拟南芥完整基因组的5条染色体上对应的“新区域”和老区域的位置分布图,其中蓝色的部分代表“老区域”,红色的部分代表“新区域”。同学们也可以根据自己想要展示的分区来进行设置数据集哦!
保存图像
现在,我们已经完成了图形的绘制。我们可以使用 R 中的任何绘图设备将图形保存到文件中。在这里,我们将使用 png 函数将图形保存为 PNG 文件。
# 输出图片png(filename = "newGeneAreas111.jpg", type = "cairo", height = 800, width = 1100)print(p)dev.off()
完整代码:
library(ggplot2)# 读取数据a <- read.table(file = "newGeneArea-15.txt", header = F, quote = "", sep = "t", stringsAsFactors = F)# 提取染色体号g <- sprintf("Chr%s", unique(a[, 4]))# 提取每段长度a[, 6] <- a[, 3] - a[, 2]# 标记染色体号for (i in 1:length(a[, 1])) {if (a[i, 4] == "chr1") {a[i, 7] = 1} else if (a[i, 4] == "chr2") {a[i, 7] = 2} else if (a[i, 4] == "chr3") {a[i, 7] = 3} else if (a[i, 4] == "chr4") {a[i, 7] = 4} else if (a[i, 4] == "chr5") {a[i, 7] = 5}}# 设置颜色colors <- c("lightsteelblue2", "orangered3")# 绘图p <- ggplot(a, aes(x = a[, 7], y = a[, 6], fill = a[, 5], group = a[, 4])) +geom_bar(stat = "identity") +theme_bw() +theme(panel.grid.major = element_blank(),legend.position = c(0.9, 0.9),legend.title = element_blank()) +scale_fill_manual(values = colors) +scale_x_continuous(breaks = c(1:5), labels = g) +labs(x = "", y = "") +scale_y_continuous(labels = c(0, "10MB", "20MB", "30MB"))# 输出图片png(filename = "newGeneAreas111.jpg", type = "cairo", height = 800, width = 1100)print(p)dev.off()svg(filename = "newGeneAreas111.svg", height = 7, width = 14)print(p)dev.off()
怎么样,今天的分区图表绘制你学会怎么用了吗!
更多ggplot绘图应用学习资源请大家移步小果专属云生信平台搜索更多资源哦!
小果专属云生信平台:云生信 – 学生物信息学 (biocloudservice.com)-文末点击阅读原文可以跳转
ggplot绘制互作网络图:http://www.biocloudservice.com/703/703.php
云生信平台也有绘图专版的学习模块哦,快来找到你想学习的专属
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

