小果带你做单基因相关miRNA的分析绘图
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

基因-微小RNA(miRNA)的相关性分析,可以探索基因和miRNA之间的潜在相互作用和调控关系。意义在于帮助研究人员分析基因和miRNA之间的关系,并找到与目标基因相关的miRNA,以及它们之间的调控机制。这对于深入理解基因调控网络、发现潜在的生物标志物研究疾病的发生机制等方面都具有重要意义。
代码中选择了一个目标基因(gene)并对其与miRNA的表达量进行相关性分析。通过计算相关系数和p值,可以确定与目标基因表达量显著相关的miRNA。下面是原始下载的基因表达量数据和mirna的原始数据。
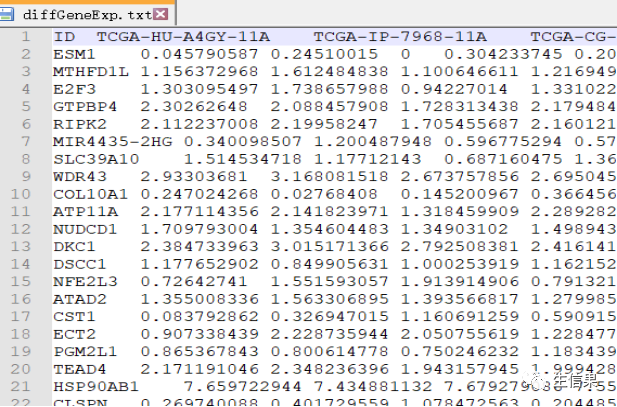
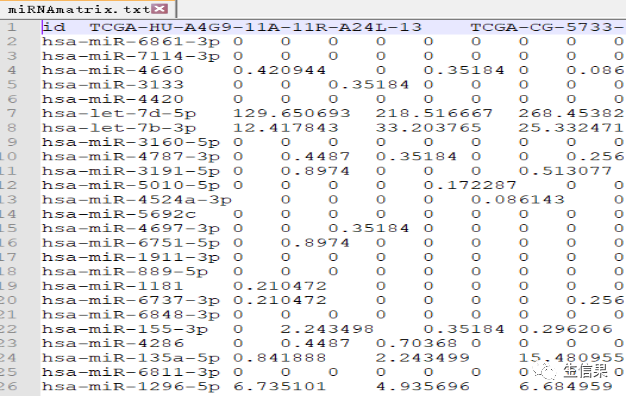
#if (!requireNamespace("BiocManager", quietly = TRUE))# install.packages("BiocManager")#BiocManager::install("limma")#install.packages("ggpubr")#install.packages("ggExtra")# 首先,检查是否已经安装了名为"BiocManager"的包,如果没有则进行安装。接下来,安#装"limma"、"ggpubr"和"ggExtra"包。#引用包library(limma)library(reshape2)library(ggpubr)library(ggExtra)corFilter=0.2 #相关系数过滤标准pvalueFilter=0.001 #pvalue过滤标准gene="VCAN" #示例展示的基因名称expFile="diffGeneExp.txt" #基因表达文件miExpFile="miRNAmatrix.txt" #miRNA表达文件mirnaFile="miRNA.txt" #miRNA列表文件,之前rownames提取出的行名

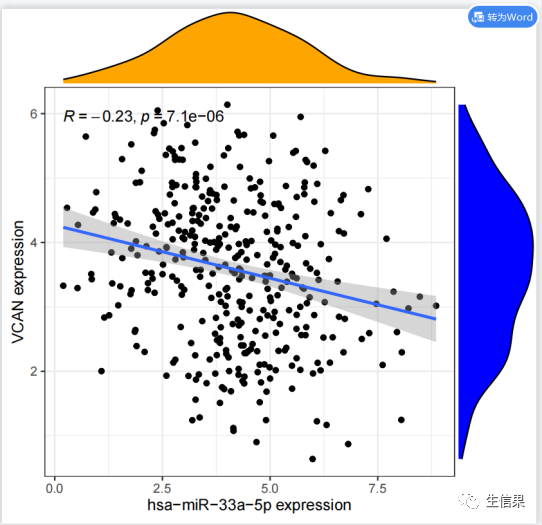
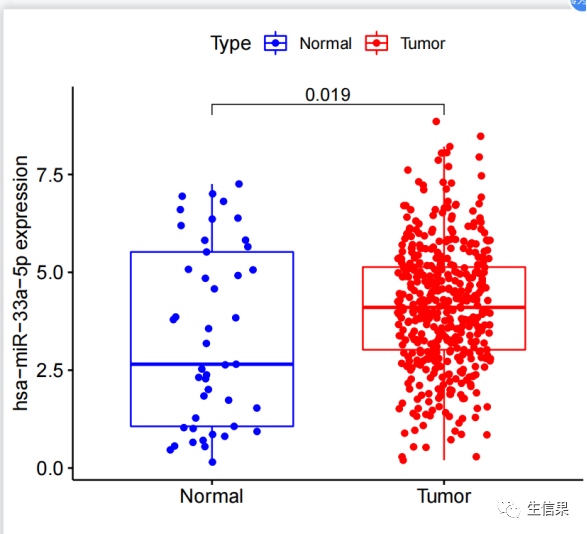
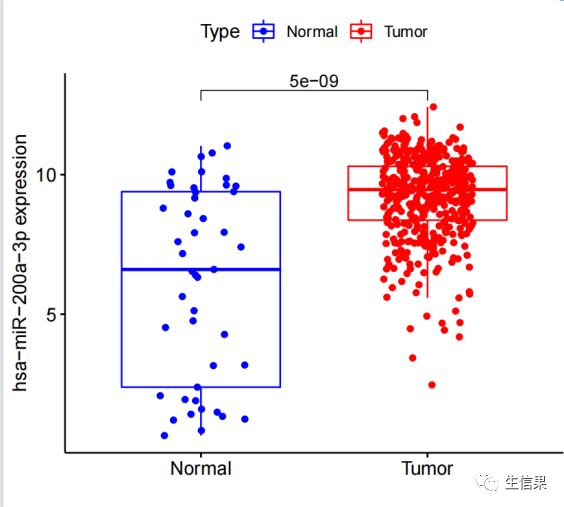
#初始化了一些变量,包括相关系数过滤阈值(corFilter)、p值过滤阈值(pvalueFilter)、#目标基因名称(gene)、基因表达文件名(expFile)、miRNA表达文件名(miExpFile)、#miRNA列表文件名(mirnaFile)#读取基因表达文件,提取目标基因的表达量rt=read.table(expFile, header=T, sep="t", check.names=F)rt=as.matrix(rt)rownames(rt)=rt[,1]exp=rt[,2:ncol(rt)]dimnames=list(rownames(exp),colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)data=avereps(data)data=data[gene,,drop=F]#从指定的文件(expFile)中读取基因表达数据。基因名称作为行名,提取目标基因的表达#量数据。将表达量数据转换为矩阵形式,存储在变量"data"中#删掉正常样品group=sapply(strsplit(colnames(data),"\-"), "[", 4)group=sapply(strsplit(group,""), "[", 1)group=gsub("2", "1", group)conData=data[,group==1,drop=F]treatData=data[,group==0,drop=F]logFC=mean(treatData)-mean(conData)geneExp=data[,group==0,drop=F]# 从基因表达数据中分离出正常样本和肿瘤样本。计算肿瘤样本和正常样本的平均表达#量之差(logFC)。将肿瘤组的基因表达数据存储在变量"geneExp"中。#读取miRNA表达数据rt1=read.table(miExpFile, header=T, sep="t", check.names=F)rt1=as.matrix(rt1)rownames(rt1)=rt1[,1]exp1=rt1[,2:ncol(rt1)]dimnames1=list(rownames(exp1),colnames(exp1))miRNA=matrix(as.numeric(as.matrix(exp1)), nrow=nrow(exp1), dimnames=dimnames1)miRNA=avereps(miRNA)miRNA=miRNA[rowMeans(miRNA)>0,]#从指定的文件(miExpFile)中读取miRNA表达数据。将miRNA名称作为行名,提取miRNA#的表达量数据。将表达量数据转换为矩阵形式,存储在变量"miRNA"中#提取目标miRNA表达量miList=read.table(mirnaFile, header=F, sep="t", check.names=F)sameMirna=intersect(row.names(miRNA), as.vector(miList[,1]))miRNA=miRNA[sameMirna,]miRNA=log2(miRNA+1)#正常和肿瘤样品数目group=sapply(strsplit(colnames(miRNA),"\-"), "[", 4)group=sapply(strsplit(group,""), "[", 1)group=gsub("2", "1", group)miRNAtumor=miRNA[,group==0]conNum=length(group[group==1]) #正常组样品数目treatNum=length(group[group==0]) #肿瘤组样品数目sampleType=c(rep("Normal",conNum), rep("Tumor",treatNum))colnames(miRNAtumor)=gsub("(.*?)\-(.*?)\-(.*?)\-(.*?)\-.*","\1\-\2\-\3-\4",colnames(miRNAtumor))#样品取交集sameSample=intersect(colnames(miRNAtumor), colnames(geneExp))miRNAtumor=miRNAtumor[,sameSample,drop=F]geneExp=geneExp[,sameSample,drop=F]y=as.numeric(geneExp[gene,])# 从miRNA和基因表达数据中获取样本的类型(正常/肿瘤)信息。获取样本的类型和#样本数目。确定miRNA和基因表达数据中样本的交集,即在两个数据中都存在的样本。#相关性检验outTab=data.frame()for(i in row.names(miRNAtumor)){if(sd(miRNAtumor[i,])>0.01){#miRNA差异分析miLogFC=mean(miRNA[i,((conNum+1):ncol(miRNA))])-mean(miRNA[i,1:conNum])test=wilcox.test(miRNA[i,] ~ sampleType)diffPvalue=test$p.value#相关性分析x=as.numeric(miRNAtumor[i,])corT=cor.test(x, y, method="spearma")cor=corT$estimatepvalue=corT$p.valueoutTab=rbind(outTab,cbind(Gene=gene, miRNA=i, cor, pvalue, logFC=miLogFC, diffPval=diffPvalue))#对满足相关性过滤条件的miRNA进行可视化if((cor< -corFilter) & (pvalue<pvalueFilter)){#绘制相关性散点图df1=as.data.frame(cbind(x,y))p1=ggplot(df1, aes(x, y)) + xlab(paste0(i, " expression")) + ylab(paste0(gene, " expression"))+geom_point() + geom_smooth(method="lm",formula = y ~ x) + theme_bw()+stat_cor(method = 'spearman', aes(x =x, y =y))p2=ggMarginal(p1, type="density", xparams=list(fill = "orange"), yparams=list(fill = "blue"))#保存相关性图形pdf(file=paste0("cor.",i,".pdf"), width=5.2, height=5)print(p2)dev.off()#miRNA差异的箱线图rt=data.frame(Expression=miRNA[i,], Type=sampleType)group=levels(factor(rt$Type))rt$Type=factor(rt$Type, levels=group)comp=combn(group,2)my_comparisons=list()for(k in 1:ncol(comp)){my_comparisons[[k]]<-comp[,k]}#绘制boxplotboxplot=ggboxplot(rt, x="Type", y="Expression", color="Type",xlab="", ylab=paste0(i, " expression"),legend.title="Type",palette = c("blue","red"),add = "jitter")+stat_compare_means(comparisons = my_comparisons)#输出箱线图pdf(file=paste0("diff.",i,".pdf"),width=5,height=4.5)print(boxplot)dev.off()}}}
对miRNA表达数据中的每个miRNA进行循环遍历:
如果该miRNA的表达量的标准差大于0.01:
计算miRNA在肿瘤组和正常组之间的平均表达量差异(miLogFC)。
进行miRNA表达量与目标基因表达量之间的相关性分析,使用的是Spearman相关系数和对应的p值。
将相关性结果存储在数据框”outTab”中。
对满足相关性过滤条件的miRNA进行可视化,包括绘制相关性散点图和差异的箱线图,并将结果保存为PDF文件。
#输出相关性结果outTab=outTab[order(as.numeric(as.vector(outTab[,"cor"]))),]write.table(file="net.network.txt",outTab,sep="t",quote=F,row.names=F)#输出相关性节点属性miNode=data.frame(Node=unique(as.vector(outTab[,"miRNA"])), Type="miRNA")geneNode=data.frame(Node=unique(as.vector(outTab[,"Gene"])), Type="Gene")nodeOut=rbind(miNode, geneNode)write.table(nodeOut, file="net.node.txt", sep="t", quote=F, row.names=F)#输出相关性显著的结果outTab=outTab[((as.numeric(as.vector(outTab[,"cor"]))<-corFilter)&(as.numeric(as.vector(outTab[,"pvalue"])) < pvalueFilter)),]write.table(file="cor.sig.txt",outTab,sep="t",quote=F,row.names=F)#输出相关性显著miRNA的表达量corMirna=unique(as.vector(outTab[,"miRNA"]))corMirnaExp=miRNA[corMirna,,drop=F]corMirnaExp=rbind(ID=colnames(corMirnaExp), corMirnaExp)write.table(corMirnaExp, file="cor.MirnaExp.txt", sep="t", quote=F, col.names=F)
通过基因和miRNA之间的相关性分析,可以找到基因和miRNA之间的调控关系。通过分析它们之间的相关性,可以识别出与目标基因表达密切相关的miRNA。这有助于理解基因调控网络中的调节模式和机制。发现潜在的miRNA调控靶点:相关性分析可以帮助确定miRNA的潜在靶点基因。如果一个miRNA与某个基因呈现显著的相关性,那么该基因很可能是该miRNA的调控靶点。
下面我们来看看结果输出文件列表和结果图示范。
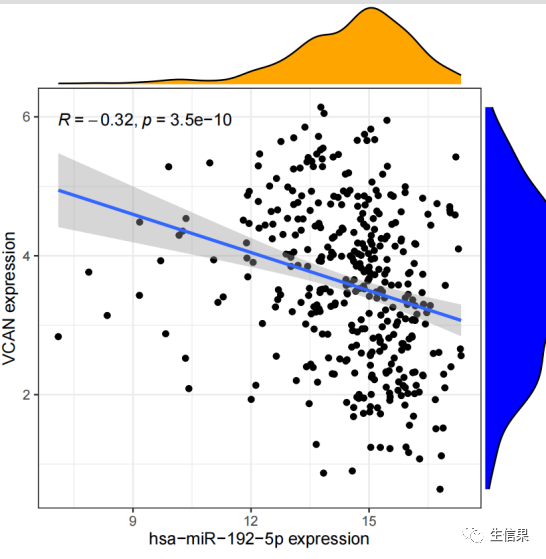
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。
其他相关分析内容,例如预测肿瘤样本药物敏感性分析(http://www.biocloudservice.com/712/712.php),预测某样本亚型对免疫治疗的反应(http://www.biocloudservice.com/292/292.php),单样本富集算法分析免疫浸润丰度(http://www.biocloudservice.com/106/106.php),计算64种免疫细胞相对含量(http://www.biocloudservice.com/107/107.php)等都可以用本公司新开发的零代码云平台生信分析小工具,一键完成该分析奥,感兴趣的小伙伴欢迎来尝试奥,网址:http://www.biocloudservice.com/home.html。今天小果的分享就到这里,下期在见奥。
点击“阅读原文”立刻拥有
↓↓↓