在使用R语言环境中如何进行正态性检验的保姆级教程
点击蓝字,关注我们
在生物信息学研究中,经常需要进行假设检验来比较不同组间的差异或者进行相关性分析。在许多假设检验中,常常要求数据满足正态性假设,如t检验、ANOVA等。因此,对生物学数据进行正态性检验是必要的,以确保所应用的统计方法的准确性和可靠性。
以下是几种常见的正态性检验方法:
1. Shapiro-Wilk检验(Shapiro-Wilk Test):
Shapiro-Wilk检验是一种经典的正态性检验方法,适用于较小样本(通常不超过5000个观测值)。该检验的原假设是数据来自正态分布。在使用Shapiro-Wilk检验时,如果p-value小于设定的显著性水平(通常为0.05),我们会拒绝原假设,即认为数据不是来自正态分布。
2. Anderson-Darling检验(Anderson-Darling Test):
Anderson-Darling检验也是一种用于正态性检验的方法,适用于较小样本。它对正态性的敏感性较高,因此在样本较小的情况下,常常比Shapiro-Wilk检验更为推荐。
3. Kolmogorov-Smirnov检验(Kolmogorov-Smirnov Test):
Kolmogorov-Smirnov检验是一种较为通用的分布拟合检验方法,可以用于检验样本是否来自特定分布(包括正态分布)。在正态性检验中,我们会使用Kolmogorov-Smirnov检验来比较样本的累积分布函数(CDF)与理论正态分布的CDF之间的差异。
4. Lilliefors检验:
Lilliefors检验是对Kolmogorov-Smirnov检验的修正,针对小样本情况下的正态性检验。它比Kolmogorov-Smirnov检验更适用于小样本数据。
5. Q-Q图(Quantile-Quantile Plot):
Q-Q图不是严格意义上的统计检验方法,而是一种可视化工具,用于检验数据是否来自某个分布(通常是正态分布)。Q-Q图绘制样本数据的分位数与理论分布的分位数之间的关系。如果数据来自正态分布,点在Q-Q图上应该大致沿着一条直线分布。
在生物信息学中,正态性检验是一种常用的统计方法,用于验证生物学数据是否近似于正态分布。正态性检验在生物信息学研究中有广泛的应用,尤其在高通量数据分析中,如基因表达数据、蛋白质组学数据和代谢组学数据等。
在基因表达研究中,我们经常需要比较不同基因在不同条件下的表达水平。对于每个基因的表达值,可以应用正态性检验来验证其是否近似于正态分布,从而确定是否可以使用t检验、方差分析等方法进行差异表达分析。
示例:
# 安装并加载 airway 包> install.packages("airway")> library(airway)# 使用 airway 数据集的 gene 表达值> gene_expression <- airway$gene# 使用 Shapiro-Wilk 正态性检验> shapiro_test_result <- shapiro.test(gene_expression)# 查看 Shapiro-Wilk 检验结果的 p-value> p_value_shapiro <- shapiro_test_result$p.value# 输出 p-value> print(p_value_shapiro)# 可视化基因表达值的分布情况> ggplot(data.frame(gene_expression), aes(x = gene_expression)) ++ geom_histogram(bins = 30) ++ theme_minimal()
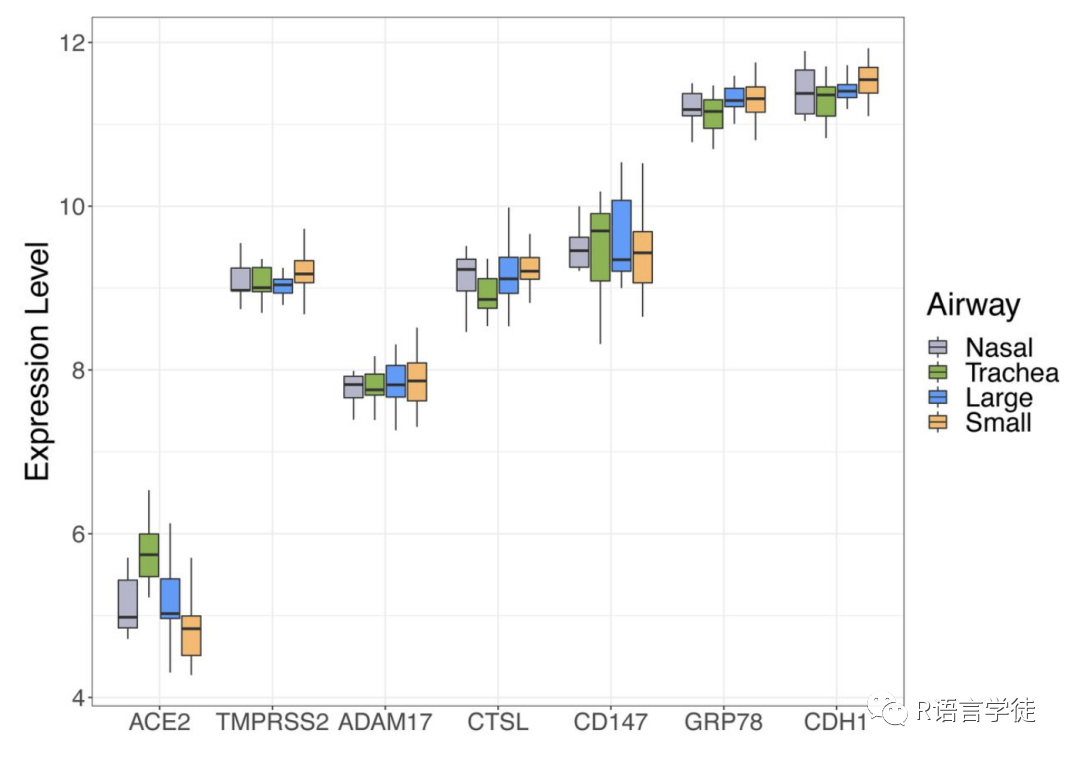
R中的airway数据集是airway包中提供的一个基因表达数据集,包含了在人支气管上皮细胞中进行生物学实验的基因表达值。在这个例子中,我们使用了airway数据集中的gene列作为基因表达值,然后执行了Shapiro-Wilk正态性检验,查看基因表达值是否满足正态分布。同时,我们使用直方图可视化了基因表达值的分布情况。
在蛋白质组学研究中,如质谱数据分析,我们可能会检测数百或数千个蛋白质的表达水平。对于这些数据,我们可以使用正态性检验来评估每个蛋白质的表达是否满足正态分布,从而为后续的差异分析和蛋白质功能富集分析提供基础。
示例:
# 安装并加载 Vim 包> install.packages("Vim")> library(Vim)# 导入 P100 数据集> data(P100)# 取出蛋白质组学数据作为基因表达值(代替基因表达数据)> protein_expression <- P100# 使用 Shapiro-Wilk 正态性检验> shapiro_test_result <- shapiro.test(protein_expression)# 查看 Shapiro-Wilk 检验结果的 p-value> p_value_shapiro <- shapiro_test_result$p.value# 输出 p-value> print(p_value_shapiro)# 可视化蛋白质组学数据的分布情况> ggplot(data.frame(protein_expression), aes(x = protein_expression)) ++ geom_histogram(bins = 30) ++ theme_minimal()

在R中,可以使用P100数据集,它包含了人类淋巴细胞系列中的蛋白质组学数据,是一个广泛用于统计和机器学习方法的示例数据集。然后执行了Shapiro-Wilk正态性检验,查看蛋白质组学数据是否满足正态分布。
代谢组学研究涉及到大量的代谢产物测量数据,通常用于识别生物样本的代谢组成。在代谢组学数据分析中,正态性检验可用于验证每个代谢产物的测量值是否符合正态分布,从而决定是否适合应用t检验或相关性分析等方法。
示例:
# 安装并加载 MetaboAnalystR 包> install.packages("MetaboAnalystR")> library(MetaboAnalystR)# 导入 ms.data 数据集> data(ms.data)# 取出代谢组学数据作为基因表达值(代替基因表达数据)> metabolomics_data <- ms.data$peakTable# 使用 Shapiro-Wilk 正态性检验> shapiro_test_result <- shapiro.test(metabolomics_data)# 查看 Shapiro-Wilk 检验结果的 p-value> p_value_shapiro <- shapiro_test_result$p.value# 输出 p-value> print(p_value_shapiro)# 可视化代谢组学数据的分布情况> ggplot(data.frame(metabolomics_data), aes(x = metabolomics_data)) ++ geom_histogram(bins = 30) ++ theme_minimal()
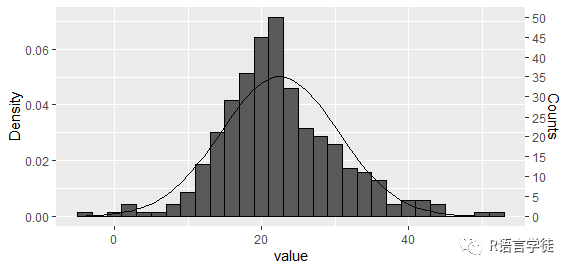
在R中,可以使用ms.data数据集,它包含了代谢组学实验中的质谱数据,是一个用于代谢组学分析的示例数据集。然后执行了Shapiro-Wilk正态性检验,查看代谢组学数据是否满足正态分布。
需要注意的是,在进行正态性检验时,样本大小对检验结果的影响较大。对于大样本,即使数据不完全符合正态分布,某些统计方法的假设条件也可能得到满足。此外,生物学数据往往受到多种因素的调控和噪声的干扰,因此即使数据不是严格的正态分布,有时也可以在合理范围内进行数据分析。
以上就是对normality test的简单介绍啦,正态性检验(normality test)是统计学中常用的一种检验方法,用于检验一个样本是否来自正态分布(即高斯分布)。在许多统计方法中,假设数据来自正态分布是一个常见的前提条件,如t检验、方差分析等。在使用这些方法之前,我们需要先验证数据是否满足正态性假设。在探索性数据分析中,了解数据的分布情况对于数据分析至关重要。正态性检验可以帮助我们确定数据是否近似于正态分布。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小师妹哦~~或者也可以关注我们的官网也会持续更新的哦~ http://www.biocloudservice.com/home.html

