小花带你学免疫浸润相关基因的Venn图绘制
点击蓝字 关注我们
Venn图可以直观地显示不同条件下的差异基因之间的重叠情况。通过交叉区域的大小和重叠区域的数量,可以了解在不同条件下具有共同表达变化的基因数量和共有的基因集合。筛选出具有共同调节的基因:通过观察Venn图中的交叉区域,可以筛选出在不同条件下具有共同调节的基因。交叉区域中的基因表示在多个条件下均发生表达变化,这些基因可能在相似的生物过程或信号通路中起着重要的调节作用。可视化和报告结果:Venn图作为差异表达分析的可视化工具,可以直观地呈现基因的重叠情况和共同调节的模式。Venn图可以帮助确定在特定条件下特异性的差异基因。通过观察Venn图中单独的区域,可以识别在某个条件下具有特定表达变化的基因,这些基因可能与该条件下的生物过程或疾病机制密切相关。
确定特定条件下的差异基因:Venn图可以帮助确定在特定条件下特异性的差异基因。通过观察Venn图中单独的区域,可以识别在某个条件下具有特定表达变化的基因,这些基因可能与该条件下的生物过程或疾病机制密切相关。通过差异表达分析结果文件,生成基质细胞和免疫细胞差异基因的Venn图,并输出交集基因的平均logFC值。
library("limma")library("pheatmap")fdrFilter=0.05 #fdr临界值logFCfilter=1 #logFC临界值scoreType="ImmuneScore" #按ImmuneScore分组inputFile="symbol.txt" #输入文件scoreFile="scores.txt" #score文件

#读取表达输入文件rt=read.table(inputFile,sep="t",header=T,check.names=F)rt=as.matrix(rt)

rownames(rt)=rt[,1]exp=rt[,2:ncol(rt)]dimnames=list(rownames(exp),colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)data=avereps(data)data=data[rowMeans(data)>0.1,]#删除正常样品group=sapply(strsplit(colnames(data),"\-"),"[",4)group=sapply(strsplit(group,""),"[",1)group=gsub("2","1",group)data=data[,group==0]#读取score文件,根据score中位值对样品分组score=read.table(scoreFile,sep="t",header=T,check.names=F)
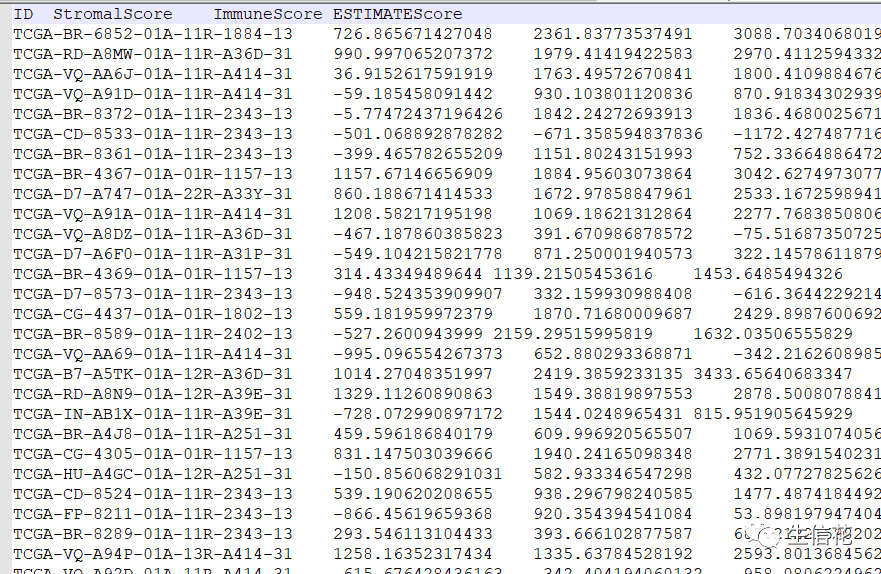
med=median(score[,scoreType])conTab=score[score[,scoreType]<=med,]treatTab=score[score[,scoreType]>med,]con=as.vector(conTab[,1])treat=as.vector(treatTab[,1])conNum=length(con)treatNum=length(treat)data=cbind(data[,con],data[,treat])#差异分析outTab=data.frame()Type=c(rep(1,conNum),rep(2,treatNum))for(i in row.names(data)){geneName=unlist(strsplit(i,"\|",))[1]geneName=gsub("\/", "_", geneName)rt=rbind(expression=data[i,],Type=Type)rt=as.matrix(t(rt))wilcoxTest<-wilcox.test(expression ~ Type, data=rt)conGeneMeans=mean(data[i,1:conNum])treatGeneMeans=mean(data[i,(conNum+1):ncol(data)])logFC=log2(treatGeneMeans)-log2(conGeneMeans)pvalue=wilcoxTest$p.valueconMed=median(data[i,1:conNum])treatMed=median(data[i,(conNum+1):ncol(data)])diffMed=treatMed-conMedif( ((logFC>0) & (diffMed>0)) | ((logFC<0) & (diffMed<0)) ){outTab=rbind(outTab,cbind(gene=i,conMean=conGeneMeans,treatMean=treatGeneMeans,logFC=logFC,pValue=pvalue))}}pValue=outTab[,"pValue"]fdr=p.adjust(as.numeric(as.vector(pValue)),method="fdr")outTab=cbind(outTab,fdr=fdr)#输出所有基因的差异情况write.table(outTab,file="Immune.all.xls",sep="t",row.names=F,quote=F)#输出差异表格diffSig=outTab[(abs(as.numeric(as.vector(outTab$logFC)))>logFCfilter& as.numeric(as.vector(outTab$fdr))write.table(diffSig,file="Immune.Diff.txt",sep="t",row.names=F,quote=F)
这样就得到了我们后续分析交集基因的Immune.Diff.txt文件,对Stromal基质细胞操作完全一样,下面是在寻找筛选关键基因的代码,我们绘制venn图简单清晰的展示。
#install.packages("VennDiagram")用于安装所需的R包VennDiagram,如果已经安装了则可以跳过安装步骤。library(VennDiagram) #引用包StromalFile="Stromal.Diff.txt" #基质细胞差异结果文件
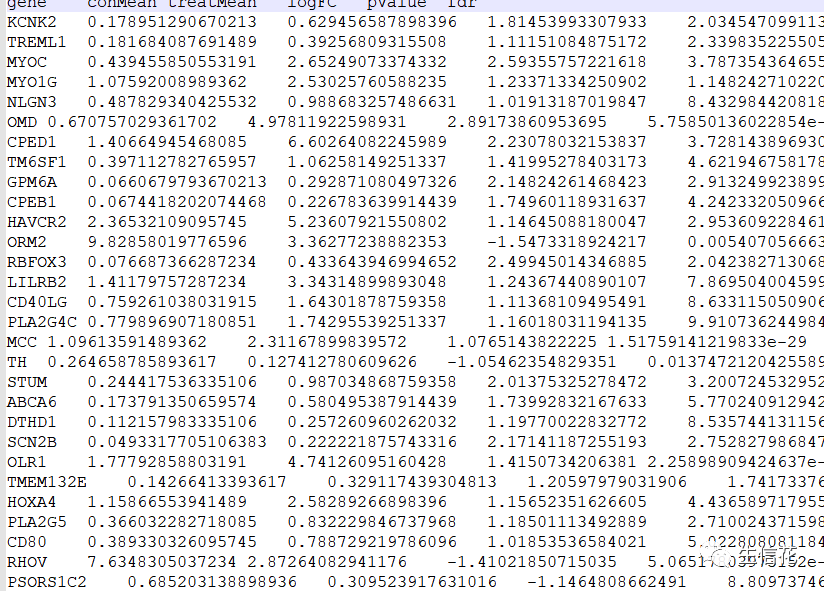
ImmuneFile="Immune.Diff.txt" #免疫细胞差异结果文件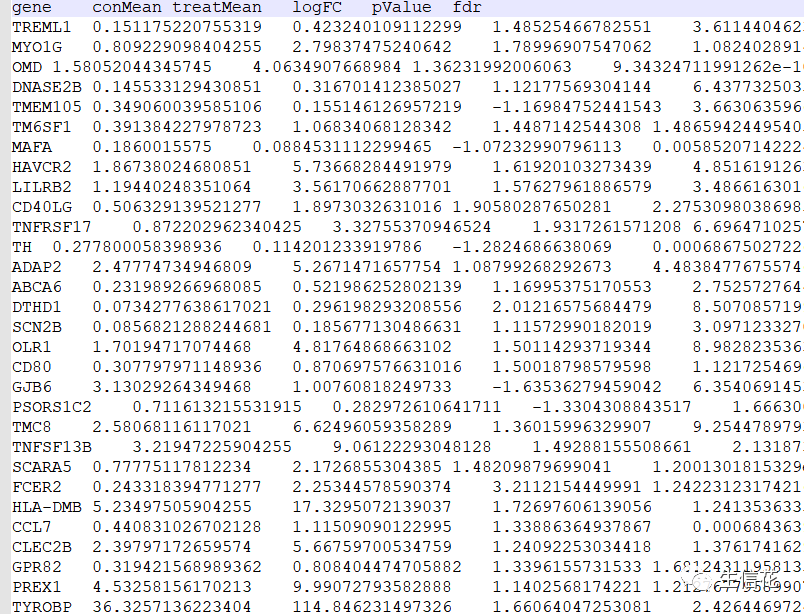
upList=list()downList=list()#这些代码指定了基质细胞差异结果文件和免疫细胞差异结果文件的文件名,#并创建了两个空的列表upList和downList,用于存储上调和下调基因的名称#读取Stromal文件Stromal=read.table(StromalFile,header=T,sep="t",check.names=F,row.names=1)upList[["Stromal"]]=row.names(Stromal[Stromal[,"logFC"]>0,])downList[["Stromal"]]=row.names(Stromal[Stromal[,"logFC"]<0,])

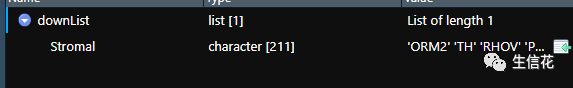
#读取基质细胞差异结果文件,并根据logFC的正负将上调和下调的基因名称#分别存储在upList[["Stromal"]]和downList[["Stromal"]]中。#读取Immune文件Immune=read.table(ImmuneFile,header=T,sep="t",check.names=F,row.names=1)upList[["Immune"]]=row.names(Immune[Immune[,"logFC"]>0,])downList[["Immune"]]=row.names(Immune[Immune[,"logFC"]<0,])

#读免疫细胞差异结果文件根据logFC的正负将上调和下调的基因名称分别存储在upList[["Immune"]]和downList[["Immune"]]中。#上调venn图upVenn=venn.diagram(upList,filename=NULL,main="Up",main.cex = 2,fill=c("darkblue", "darkgreen"),cat.cex=1.2)pdf(file="UP.venn.pdf",width=7,height=7)grid.draw(upVenn)dev.off()upGenes=Reduce(intersect,upList)#生成上调基因的Venn图。使用venn.diagram函数根据上调基因的列表upList#绘制Venn图,并设置图的标题、主标题字体大小、填充颜色等。然后使用pdf #函数创建一个输出文件,将Venn图保存为PDF格式。接下来使用grid.draw函#数绘制Venn图,并使用dev.off函数关闭PDF设备。#最后,使用Reduce函数计算上调基因列表的交集,并将结果存储在upGenes。#下调venn图downVenn=venn.diagram(downList,filename=NULL,main="Down",main.cex = 2,fill=c("darkblue", "darkgreen"),cat.cex=1.2)pdf(file="DOWN.venn.pdf",width=7,height=7)grid.draw(downVenn)dev.off()downGenes=Reduce(intersect,downList)生成下调基因的Venn图,过程与上调基因的Venn图类似,只是标题和输出文件名不同,并将结果存储在downGenes中。#输出交集基因upLogFC=(as.matrix(Stromal)[upGenes,"logFC"]+as.matrix(Immune)[upGenes,"logFC"])/2downLogFC=(as.matrix(Stromal)[downGenes,"logFC"]+as.matrix(Immune)[downGenes,"logFC"])/2logFC=c(upLogFC,downLogFC)outTab=as.data.frame(logFC)outTab=cbind(ID=row.names(outTab),outTab)colnames(outTab)=c("Gene","logFC")write.table(file="intersectGenes.txt",outTab,sep="t",quote=F,row.names=F)
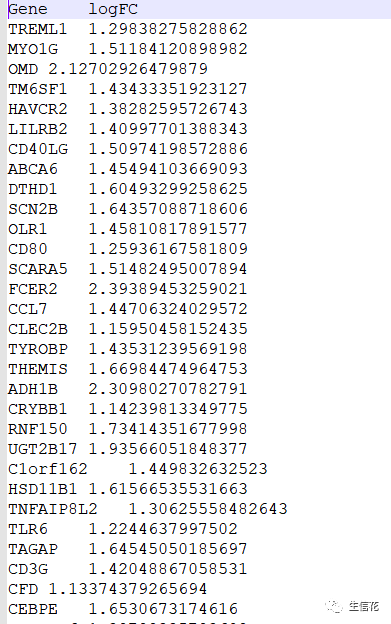
#计算交集基因的平均logFC值,并将结果存储在logFC变量中。#然后,将交集基因的ID和logFC值组合成一个数据框outTab,并为列命名。#最后,使用write.table函数将结果写入名为"intersectGenes.txt"的文件中
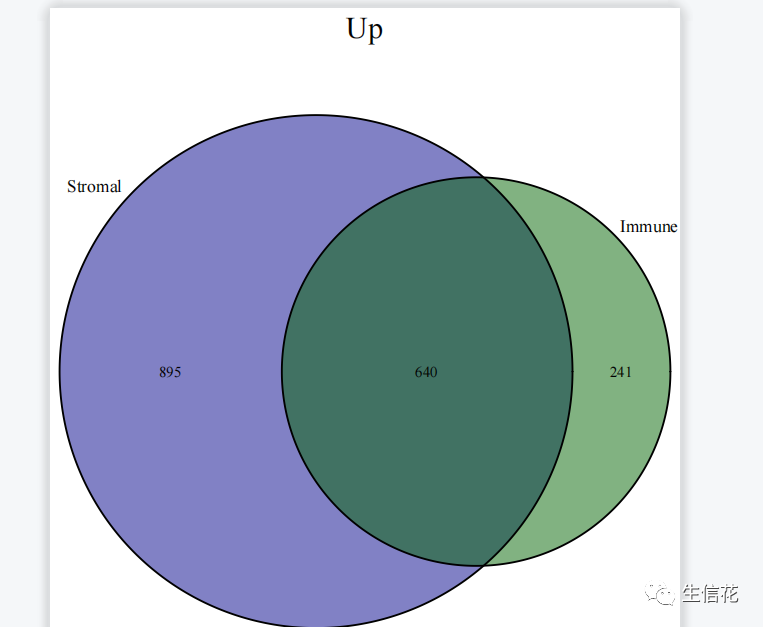
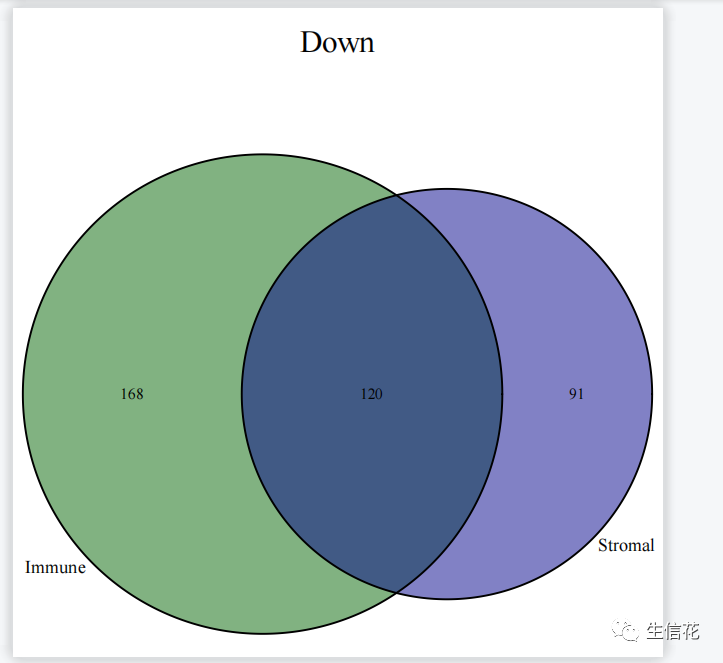
综上所述,差异表达分析的Venn图具有显示差异基因的重叠情况、筛选共同调节的基因、确定特定条件下的差异基因、辅助生物学解释和实验设计,以及可视化和报告结果的功能和意义。它在差异表达分析和生物学研究中发挥重要作用,帮助揭示基因调控和生物过程的差异,为疾病机制和治疗靶点的研究提供重要线索。下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)


![]() 点一下阅读原文了解更多资讯
点一下阅读原文了解更多资讯
