什么聚类分析一行代码就解决?说的就是K-means!
{ 点击蓝字,关注我们 }
今天小师妹给大家带来的是K-means聚类算法的教程~K-means聚类算法是一种无监督学习方法,用于将数据点分为多个不同的簇。K-means聚类算法在生物信息数据分析中有着广泛的应用。其中,基因表达聚类是最为常见的应用之一。通过将基因表达数据分为不同的簇,可以快速地了解基因的生物学特征和表达模式。蛋白质结构分类也是K-means算法的重要应用之一,通过对蛋白质结构进行分类,可以更好地理解蛋白质结构的相似性和差异性,从而为蛋白质功能研究和药物设计提供重要信息。此外,K-means算法还可以用于生物序列的模式识别,例如将DNA序列或蛋白质序列分为不同的簇,以推断它们可能具有的生物学功能。在使用K-means算法时,需要特别注意聚类结果的准确性和稳定性,需要根据具体的数据类型和研究问题进行调整和优化。
在K-means算法中,簇的数量K是一个需要预先指定的参数。算法的目标是将所有数据点分配到K个簇中,并使每个簇内部的数据点之间的距离尽可能小,而不同簇之间的距离尽可能大。
K-means算法的主要步骤包括:
step 1. 随机选择K个初始聚类中心。
step 2. 将每个数据点分配到距离最近的聚类中心所在的簇。
step 3. 对于每个簇,重新计算该簇的中心。
step 4. 重复步骤2和步骤3,直到聚类中心不再发生变化或者达到预定的迭代次数。
means算法的优点包括简单易行、可扩展性强、计算效率高等。它可以用于不同领域的数据分析和挖掘,例如图像分割、文本聚类、生物信息学数据分析等。但是,K-means算法也有一些缺点,例如需要预先指定聚类数量K、对初始聚类中心的选择敏感、容易陷入局部最优解等。因此,在使用K-means算法时,需要根据实际应用场景进行参数调整和优化,以获得更好的聚类结果。
在K-means聚类中,如何选择最优的K值是一个很重要的问题。以下是一些常用的方法:
肘部法则(Elbow method):计算不同K值下的SSE(不同聚类数量下数据点到其所属聚类中心距离的平方和)并绘制成K的函数图。在图表中,选择一个K值,使得增加K值对SSE的减少效果逐渐减弱,形成一个像手肘的拐点。这个拐点对应的K值就是最优的K值。可以使用kmeans函数的输出结果中的total.withinss参数来计算SSE。
轮廓系数(Silhouette Coefficient):计算不同K值下的轮廓系数并绘制成K的函数图。轮廓系数是一种度量数据点聚类质量的指标,其值介于-1到1之间。在图表中,选择一个K值,使得轮廓系数最大。可以使用cluster::silhouette函数来计算轮廓系数。
Gap统计量(Gap statistic):计算不同K值下的Gap统计量并绘制成K的函数图。Gap统计量是一种度量数据点聚类质量的指标,其计算方法是将数据集随机化多次,并计算随机化数据集与原始数据集之间的距离差异。在图表中,选择一个K值,使得Gap统计量最大。可以使用cluster::clusGap函数计算Gap统计量。
需要注意的是,这三种方法都存在一定的局限性,不一定适用于所有数据集。因此,最终的K值选择应该基于具体的应用场景和数据特点。
接下来,小师妹将以组织基因表达数据为例,给大家带来K-means在R语言中的具体操作过程~
首先我们需要选择一个合适的K值,这里小师妹会带大家把3种方法都过一遍哦~
我们先导入相关的R包以及我们的数据:
library(ggplot2)library(cluster)library(tissuesGeneExpression)data(tissuesGeneExpression)
肘部法则(Elbow method)
# 计算不同K值下的SSEsse <- c()for (k in 1:10) {kmeans_fit <- kmeans(e, k)sse[k] <- kmeans_fit$tot.withinss}# 绘制SSE-K曲线df <- data.frame(k = 1:10, SSE = sse)ggplot(df, aes(x = k, y = SSE)) + geom_line() + geom_point() +labs(title = "SSE-K Curve", x = "K", y = "SSE")
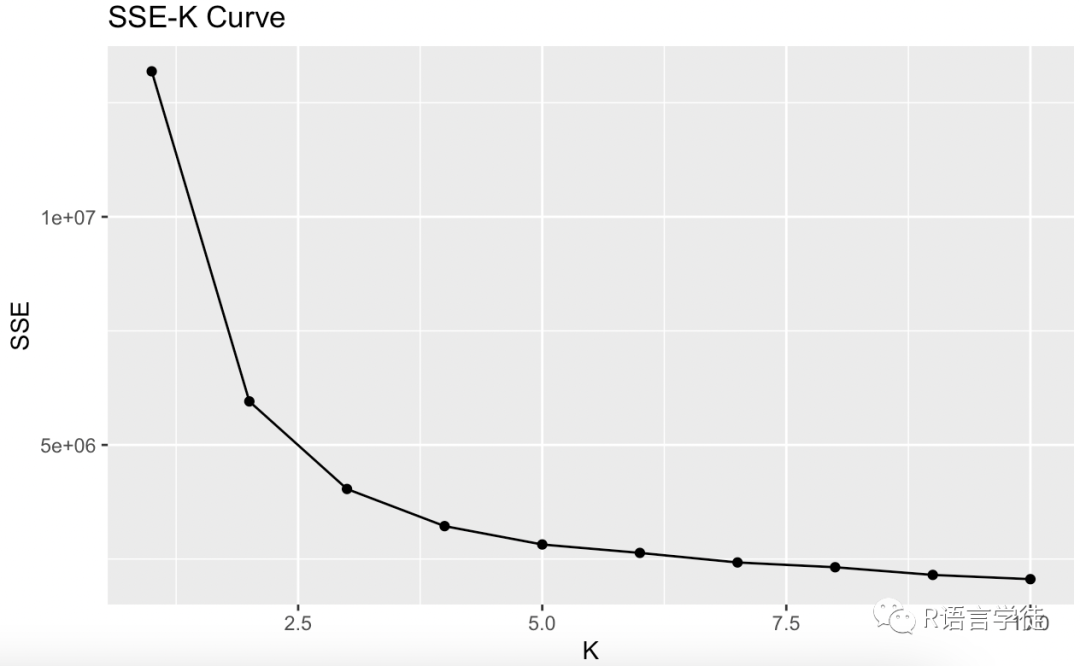
这里我们可以看到5是比较合适的分类数量
轮廓系数(Silhouette Coefficient)
# 计算不同K值下的轮廓系数sil <- c()for (k in 1:4) {kmeans_fit <- kmeans(e, k)if (nrow(table(kmeans_fit$cluster)) > 1) {sil[k] <- mean(silhouette(kmeans_fit$cluster, dist(e)))} else {sil[k] <- NA}}# 绘制轮廓系数-K曲线df <- data.frame(k = 1:4, Silhouette = sil[1:4])ggplot(df, aes(x = k, y = Silhouette)) + geom_line() + geom_point() +labs(title = "Silhouette-K Curve", x = "K", y = "Silhouette")
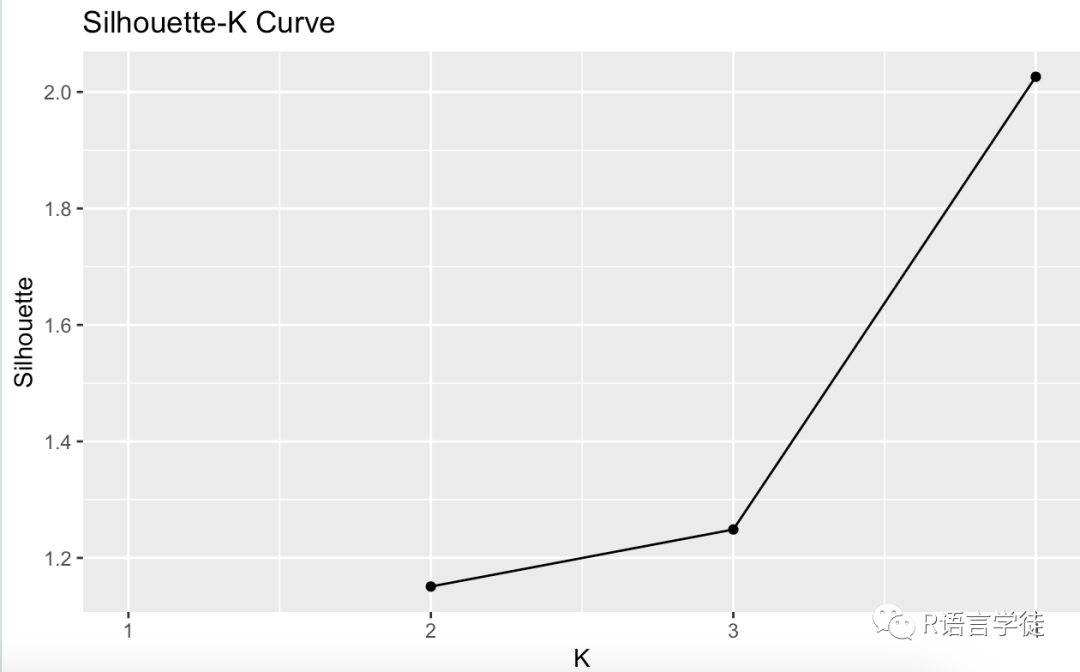
这里则建议分类数量为4
Gap统计量(Gap statistic)
# 计算不同K值下的Gap统计量gap <- clusGap(e, FUN = kmeans, nstart = 25, K.max = 10, B = 50)# 绘制Gap-K曲线df <- data.frame(k = 1:10, Gap = gap$Tab[, "gap"], SE = gap$Tab[, "SE.sim"],Upper = gap$Tab[, "gap"] + gap$Tab[, "SE.sim"],Lower = gap$Tab[, "gap"] - gap$Tab[, "SE.sim"])ggplot(df, aes(x = k)) + geom_line(aes(y = Gap)) + geom_point(aes(y = Gap)) +geom_errorbar(aes(ymin = Lower, ymax = Upper), width = 0.2) +labs(title = "Gap-K Curve", x = "K", y = "Gap")
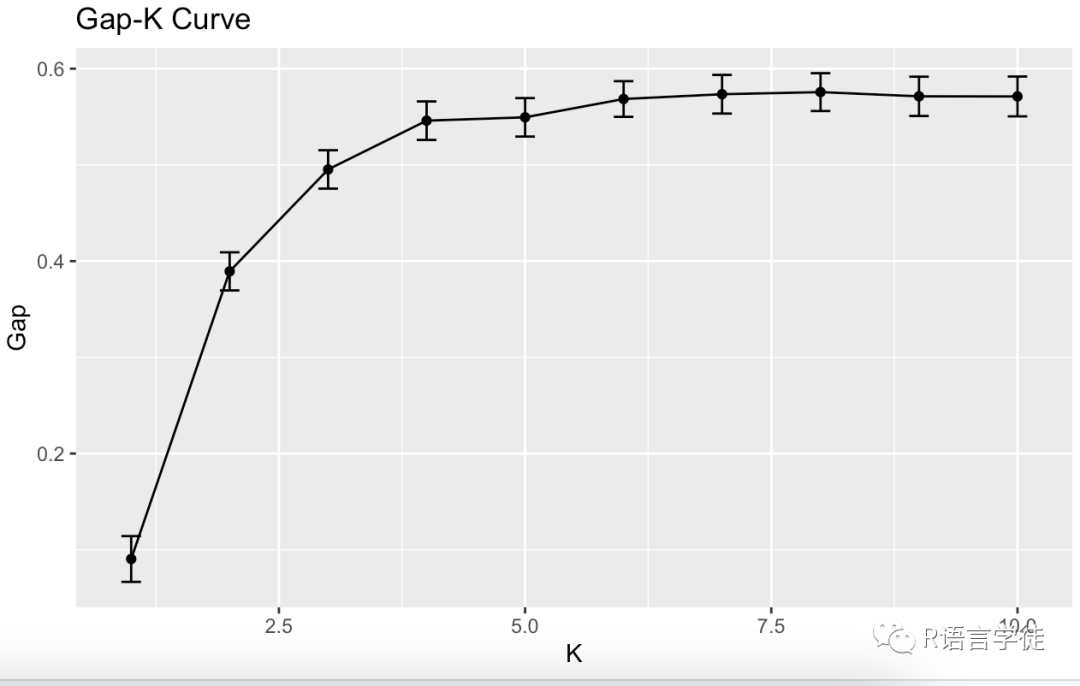
接下来我们就可以简单地使用kmeans( )函数进行聚类啦
k <- kmeans(e, 4)直接查看聚类结果
print(k$cluster)
可视化聚类结果
plot(e, col = k$cluster)绘制聚类中心
points(k$centers, col = 1:3, pch = 8, cex = 2)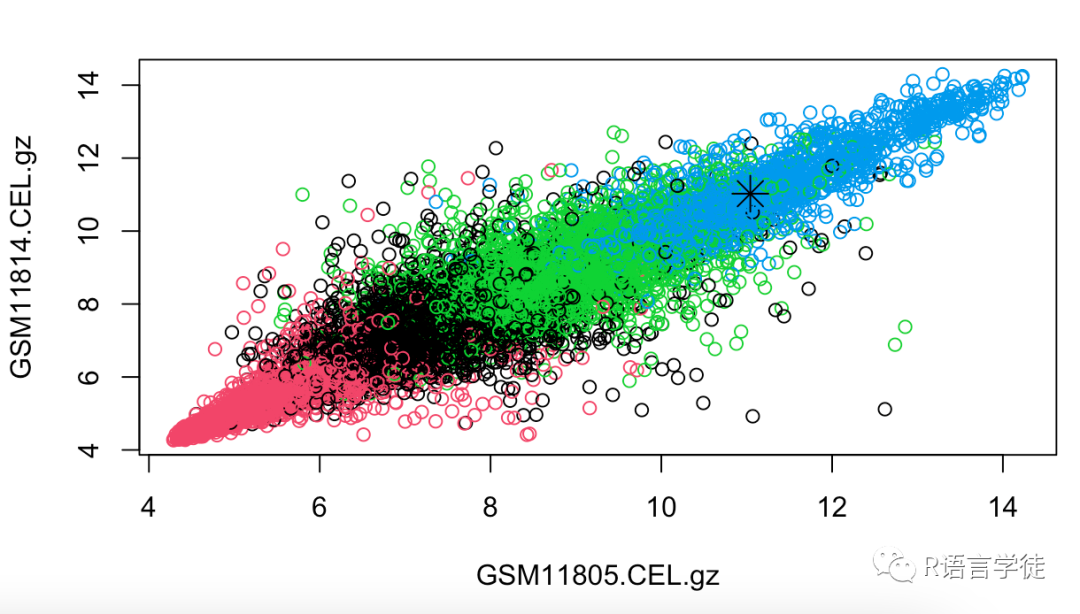
这就是K-means聚类分析啦!最重要的部分还是如何选择K值,建议小伙伴们认真思考文中关于选择K值的部分哦!
更多实用方便的小工具在云生信平台等着大家哦!
http://www.biocloudservice.com/home.html
★

