超详细的R中进行GSEA分析的流程!一看就会!


所需R包均可以用BiocManager包安装,如果没有BiocManager包,可以用以下命令安装。
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager") #安装BiocManager包BiocManager::install("clusterProfiler") #安装分析所需R包library(ReactomePA) #加载所需R包library(tidyverse)library(data.table)library(org.Hs.eg.db)library(clusterProfiler)library(biomaRt)library(enrichplot)
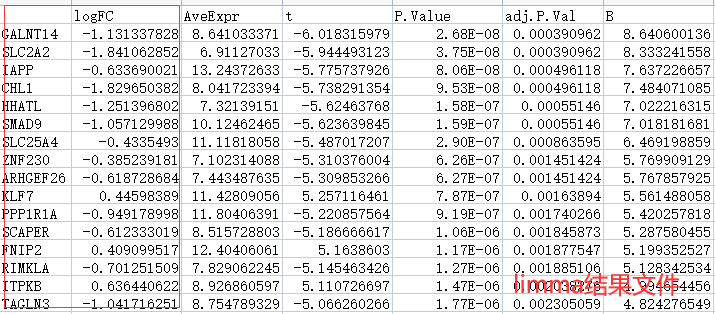
setwd("G:/job/job1/GSEA/") #进入存放数据的工作目录genelist_input <- fread(file="GSEA.txt", header = T, sep='t', data.table = F) #导入数据genename <- as.character(genelist_input[,1]) #提取第一列基因名gene_map <- select(org.Hs.eg.db, keys=genename, keytype="SYMBOL", columns=c("ENTREZID")) #将SYMBOL格式的ID换成ENTREZ格式的ID。
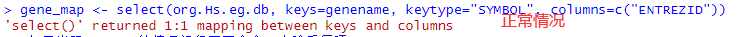
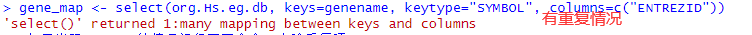
non_duplicates_idx <- which(duplicated(gene_map$SYMBOL) == FALSE)gene_map <- gene_map[non_duplicates_idx, ] #去除重复值colnames(gene_map)[1]<-"Gene" #输出mapping结果
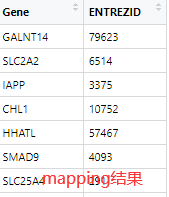
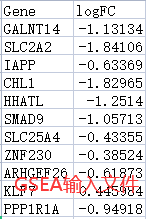
#将ENTREZID与logFC对应起来,并根据logFC的值降序排列,最终生成结果如图所示。temp<-inner_join(gene_map,genelist_input,by = "Gene")temp<-temp[,-1]temp<-na.omit(temp)temp$logFC<-sort(temp$logFC,decreasing = T)
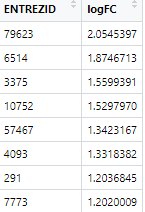
#最后我们将文件内容整理成GSEA分析所需的格式(如图所示),我们就可以开始GSEA分析了!geneList = temp[,2]names(geneList) = as.character(temp[,1])geneList

Go_gseresult <- gseGO(geneList, 'org.Hs.eg.db', keyType = "ENTREZID", ont="BP", pvalueCutoff=0.05) #使用GSEA进行GO富集分析('org.Hs.eg.db':对应物种的数据库;ont:选择输出条目,可选“BP,MF,CC或者ALL”,pvalueCutoff:设置P的阈值)KEGG_gseresult <- gseKEGG(geneList, pvalueCutoff=0.05) #使用GSEA进行KEGG富集分析#保存富集分析结果go_results<-as.data.frame(Go_gseresult)kegg_results<-as.data.frame(KEGG_gseresult)write.csv (go_results, file ="Go_gseresult.csv")write.csv (kegg_results, file ="KEGG_gseresult.csv")
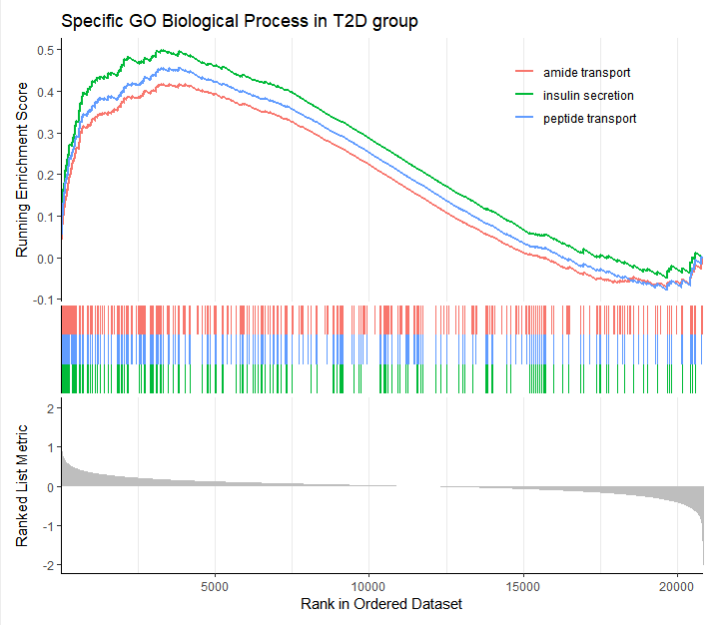
gseaplot2(Go_gseresult, 1:3, title = "Specific GO Biological Process in T2D group", pvalue_table = FALSE) #1:3:这表示在图上显示前3条富集结果,也可以根据自己分析需要指定输出某一条结果;Go_gseresult:GO富集分析结果;title:加上标题;pvalue_table:是否在图上显示P值列表。


往期推荐