5分钟学会绘制热图pheatmap
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

热图在个组学中的应用十分的广泛,在文献中经常出现,利用颜色直接展示在不同样本中展示基因、代谢物、物种等表达的水平。
一张热图像五颜六色的瓷砖一样铺在整张图中,给人视觉上的美感。通过R做heatmap是常用的方法,那我们今天就来学习一下怎样快速绘制热图吧~
安装并加载pheatmap包
#安装pheatmap包install.packages("pheatmap")#加载library(pheatmap)#设置工作路径setwd("/Users/sansan/Desktop/未命名文件夹/")
构建测试数据集
#首先构建一个数据集,作为我们学习的示例
test <- matrix(rnorm(100),10,10)test[1:10,seq(1,10,2)] <- test[1:10,seq(1,10,2)]+3test[5:10,seq(1,10,2)] <- test[5:10,seq(1,10,2)]+3colnames(test) <- paste("Sample",1:10,sep = "")rownames(test) <- paste("Gene",1:10,sep = "")test
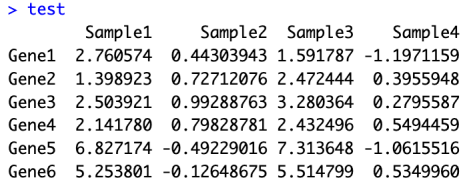
绘制简单的热图
归一化处理的必要性:是一种数据预处理中特征缩放的(feature scaling)的方法,可以让所有的数据处于一种量纲级别进行分析,更容易直观的阐明数据的差异性。例如归一化到【1,-1】的范围内,消除了异常样本导致结果不良的影响。
在应用前,首先需要了解一些必要的参数。
# clustering_method参数设定不同聚类方法,默认为"complete",可以设定为ward, ward.D, ward.D2, single, complete, average, mcquitty, median or centroid# clustering_distance_rows = "correlation"参数设定行聚类距离方法为corralation,默认为欧氏距离"euclidean"# legend_breaks参数设定图例显示范围,legend_labels参数添加图例标签legend_labels = c("1.0","2.0","3.0","4.0","5.0")#border=FALSE参数去掉边框线#border_color参数设定每个热图格子的边框色#show_rownames和show_colnames参数设定是否显示行名和列名#treeheight_row和treeheight_col参数设定行和列聚类树的高度,默认为50#display_numbers = TRUE参数设定在每个热图格子中显示相应的数值#number_color参数设置数值字体的颜色# cellwidth和cellheight参数设定每个热图格子的宽度和高度,main参数添加主标题
了解后来简单绘制一个尝试一下:
pheatmap(test,scale = "row",clustering_method = "average",clustering_distance_rows = "correlation",border=FALSE,treeheight_row = 50,treeheight_col =50,cellwidt = 15,cellheight = 20)这里用到的参数,如果你不明白意义记得去上文中寻找。
添加行信息/列信息
(用于代谢通路分析等)
在分析过程中通常会在行或者列添加一行属于
#先构建一个行的信息(用于一些代谢通路富集分析)annotation_row <- data.frame(Path = factor(rep(c("one","two"),5)))rownames(annotation_row) <- paste("Gene",1:10,sep = "")# Path#Gene1 one#Gene2 two#Gene3 one#Gene4 two#Gene5 one#Gene6 two#Gene7 one#Gene8 two#Gene9 one#Gene10 twopheatmap(test, annotation_row = annotation_row)#创建一个行的色块ann_col = list(Path = c(one = "red",two = "yellow"))#把色块映射到图上pheatmap(test, annotation_row = annotation_row, annotation_colors = ann_col)
其它一些操作
# gaps_row = c(3, 5)参数在第3和5行处添加gap, 要求对行不进行聚类#cutree_col = 2参数将列按聚类树的结果分成两部分, 要求对列进行聚类#对行和列都不聚类,自定义划分行和列的gap#labels_row 参数设置行标签pheatmap(test, annotation_col = annotation_col, cluster_rows = FALSE, cluster_cols = FALSE, gaps_row = c(3,6), gaps_col = c(2, 5),labels_row = c())
在做图的过程中我们应该充分考虑你想展示的内容是什么,要传达怎样的生物学内容,告诉读者你的研究结果,而不是一味的只追求好看。在作图的过程中你需要什么就去搜索和探索什么,这里只是简单的热图绘制方法,但是具体怎样呈现还需要进一步的学习。
此外还可以寻找很多小工具绘制热图例如:http://www.biocloudservice.com/747/747.php
今天的分享就到这里叭,小伙伴们如果有什么问题就和小果讨论吧~

点击“阅读原文”立刻拥有
↓↓↓