小果带你玩转多变量数据:如何巧用corrplot包解析各变量间的相关性

Hello,各位小伙伴大家好,很开心,小果又如期和大家见面了哦,不知道大家有没有遇到过这样的问题:我们在做实验或数据分析的时候设置了多个变量,突然,灵感迸发,想要看一下是否这几个变量之间也存在什么关系呢?这时大家应该如何处理,要怎么才能谁和谁的关系是正相关,或者哪两个变量之间是负相关呢?今天各位小伙伴可真是看对了,小果告诉大家,在R语言中的确有一个函数能计算各变量之间的相关性系数— cor()。
cor()函数可以非常简便的计算出各连续变量之间的相关系数,但当变量非常多时返回的相关系数一定会让各位小伙伴看花了眼。因此我们迫不及待的需要一个能将相关性矩阵可视化的方法,虽然有很多方法能进行相关性矩阵的可视化,且基础绘图也能完成,但在我心目中,最好用的一定还是corrplot包。
撇开它是国产的R包外,本身它的使用就非常简便,并能按照自己的审美对其可视化方法、图形布局、颜色、图例、文本标签等处进行自定义绘制。另外,虽然该包提供了约50个参数,但是常用的也就10个左右,非常适用于刚上手的小伙伴哦!
且corrplot包在生物信息学中的使用频率也很高哦:比如在基因表达和蛋白质组学的研究中,corrplot包既能帮助大家寻找到有相同表达模式的基因,助力我们对研究对象的某种分子机制的解析;也能帮助各位小伙伴可视化蛋白质之间的相互关系,帮助分析网络的模式和结构,以此来简化各位的研究哦。
总的来说,corrplot 包在生物信息学中提供了一个强大而灵活的工具,可以通过直观可视化的方式帮助大家理解生物分子之间的关系,进而从大规模数据中提取有意义的信息。说到大规模数据,不知道大家都用什么软件或者系统进行处理,小果这里呢推荐大家使用Linux系统哦,该系统界面干净,命令简单,有基因组、转录组等测序数据的处理极其友好,如果大家没有的话,可以练习效果进行租赁哦。
公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240122

#corrplot的安装与调用install.packages('corrplot')library(corrplot)library(tidyverse)library(RColorBrewer)
#读入数据data <- read.table('输入数据.txt')
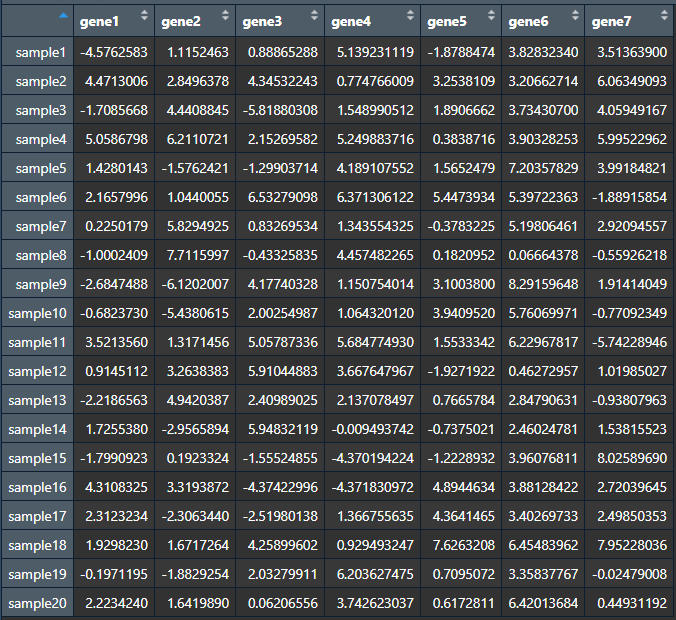
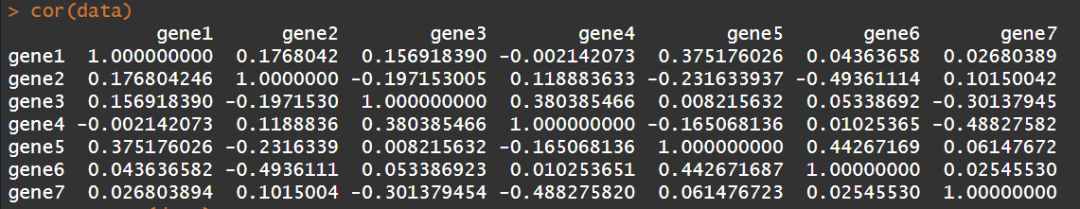
#对以上数据进行相关性分析cor <- cor(data)cor
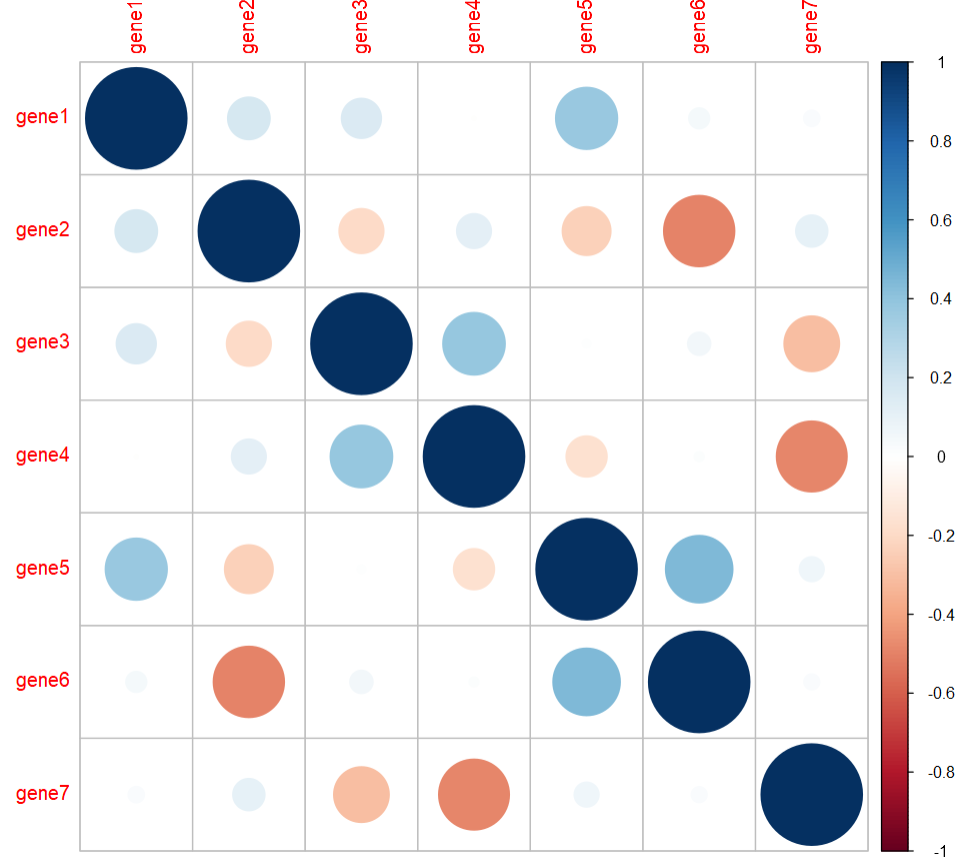
#简单出图P1 <- corrplot(cor)
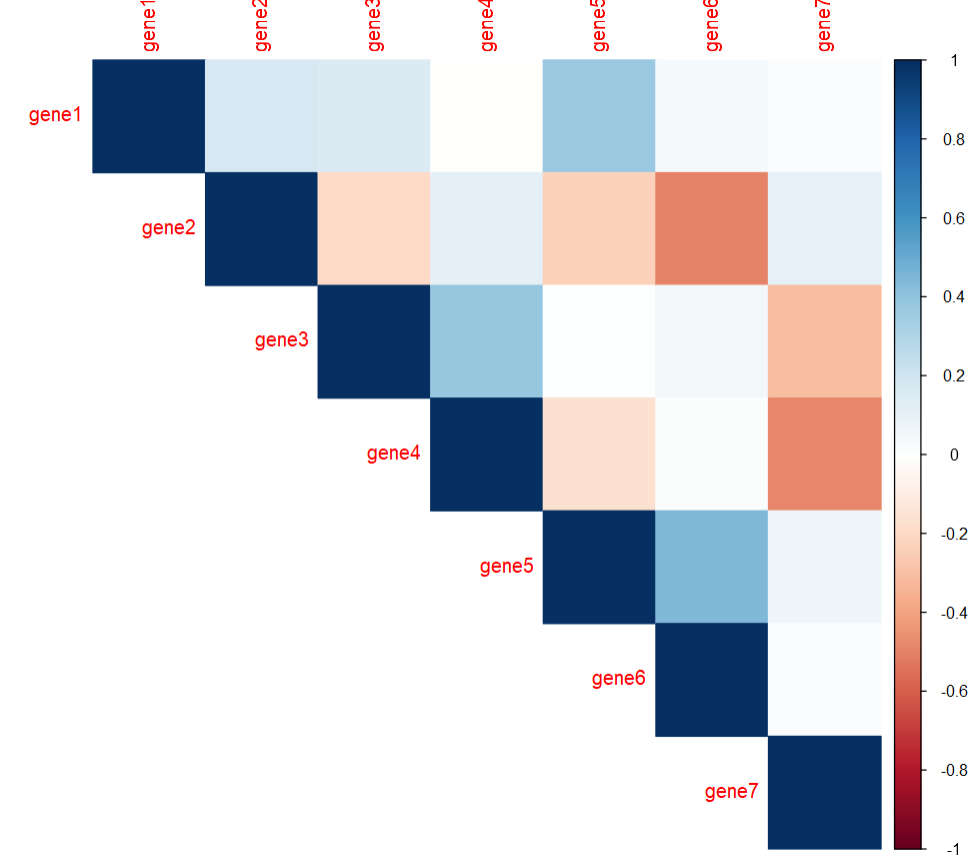
#改变展示形状P2 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper')
#改变颜色col <- colorRampPalette(c("blue", "red"))(20)P3 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper',col = col, #改变颜色,# bg = 'black' , #改变背景色title = 'test for corrplot',is.corr = T , #输入是否为相关矩阵,一般都是Tmar = c(5,5,5,5)) #与上下左右的距离
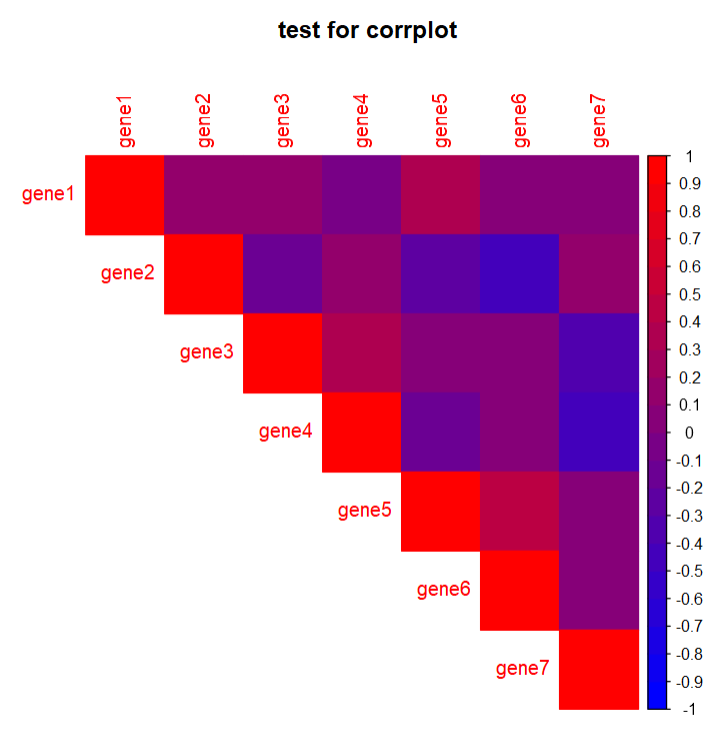
#改变边框颜色并添加相关性系数P4 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper',col = col, #改变颜色,# bg = 'black' , #改变背景色title = 'test for corrplot',is.corr = T , #输入是否为相关矩阵,一般都是T#add = T , #可通过改变type合并两种类型的图diag = F, #是否展示对角线结果outline = T, #是否加外框线addgrid.col = 'black', #改变外框线的颜色addCoef.col = 'grey', #改变添加数字的颜色addCoefasPercent = T, #改变填充数据形式,成为百分比形式mar = c(5,5,5,5), #与上下左右的距离#对图形进行排序"original", "AOE", "FPC", "hclust", "alphabet"order = c('AOE'),#层次聚类的方法"complete", "ward", "ward.D", "ward.D2", "single", "average","mcquitty", "median", "centroid"hclust.method = c('median'))
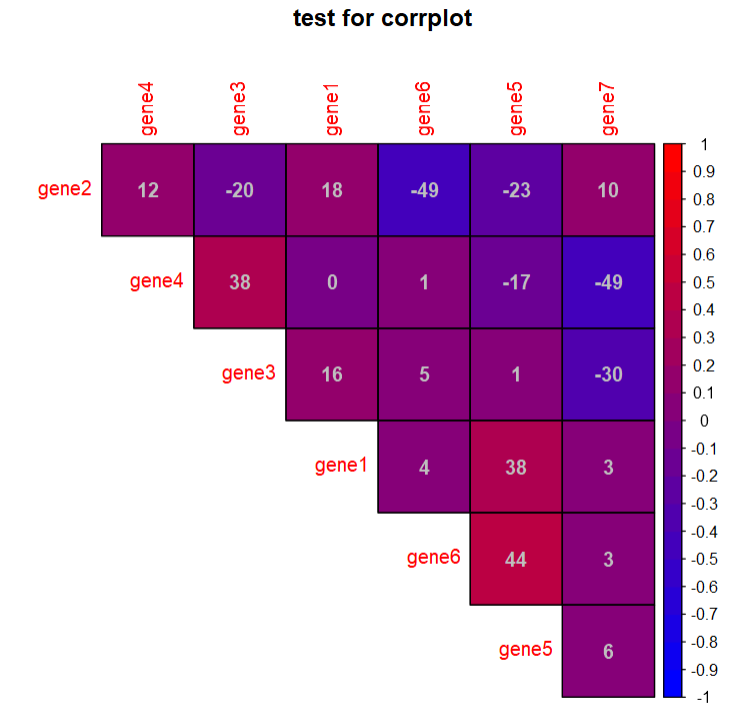
#改变行列名的表现方式,以更好的拼图P5 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper',col = col, #改变颜色,# bg = 'black' , #改变背景色title = 'test for corrplot',is.corr = T , #输入是否为相关矩阵,一般都是T#add = T , #可通过改变type合并两种类型的图diag = F, #是否展示对角线结果outline = T, #是否加外框线addgrid.col = 'black', #改变外框线的颜色addCoef.col = 'grey', #改变添加数字的颜色addCoefasPercent = T, #改变填充数据形式,成为百分比形式mar = c(5,5,5,5), #与上下左右的距离#对图形进行排序"original", "AOE", "FPC", "hclust", "alphabet"order = c('hclust'),#层次聚类的方法"complete", "ward", "ward.D", "ward.D2", "single", "average","mcquitty", "median", "centroid"hclust.method = c('median'),#改变文本标签的位置'lt', 'ld', 'td', 'd' or 'n'. 'lt'tl.pos = 'dt', #可用于两个图拼接时使用tl.cex = 0.8,tl.col = 'black' #文本大小和颜色)
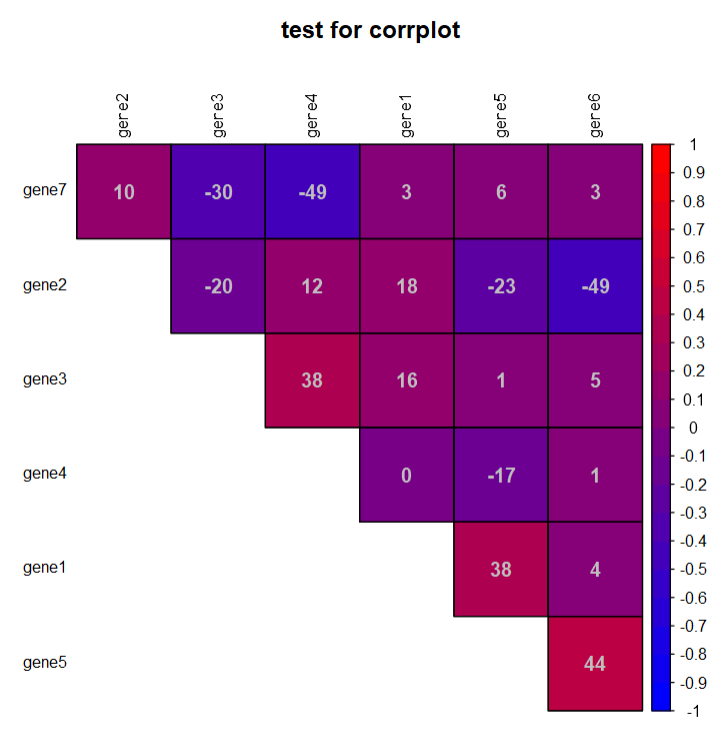
#对图例进行调整P6 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper',cl.pos = 'r', #'r''b''n'的图例位置cl.cex = 0.5 , cl.ratio = 0.3, #图例大小和宽度cl.length = 7 , #标签个数cl.align.text = 'r', #标签文本对齐方式l,r,c三种cl.offset = 0.1 #标签与图例的偏移量)
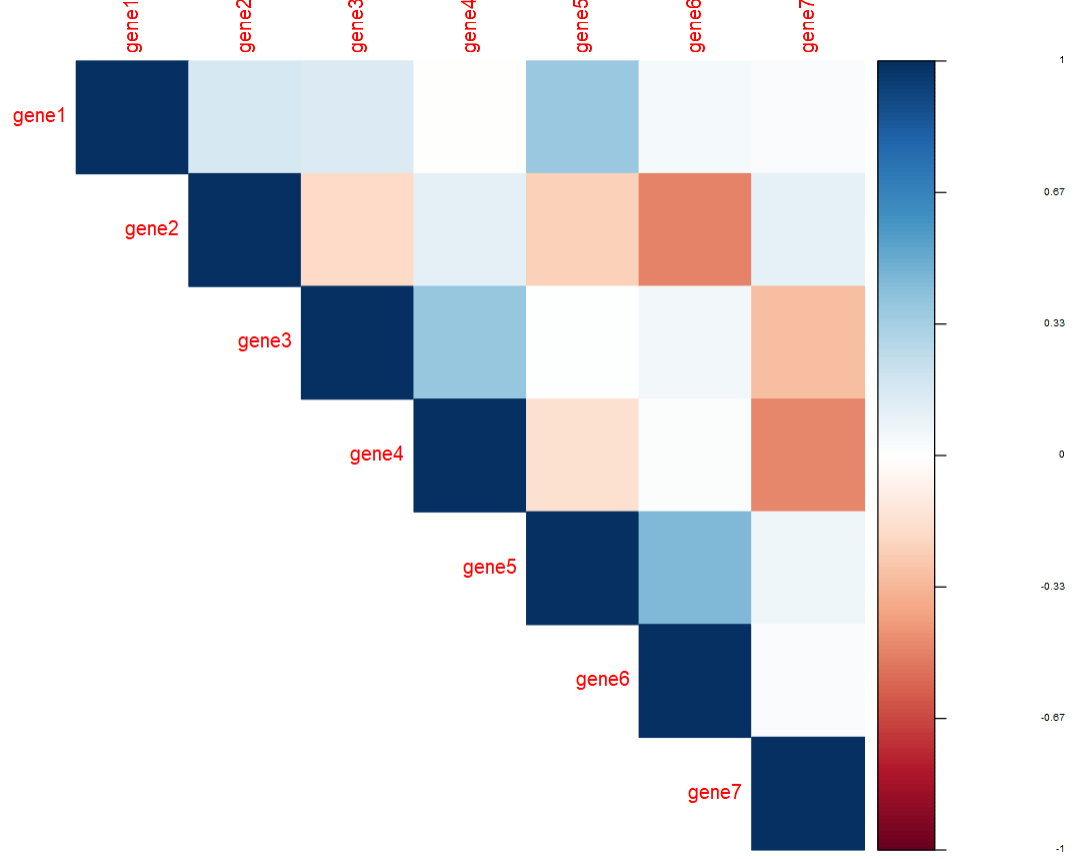
#对先的位置进行调整P7 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'shade',#展示类型"full", "lower", "upper"type = 'upper',addshade = 'positive' #在shade下有用'negative', 'positive' or 'all)
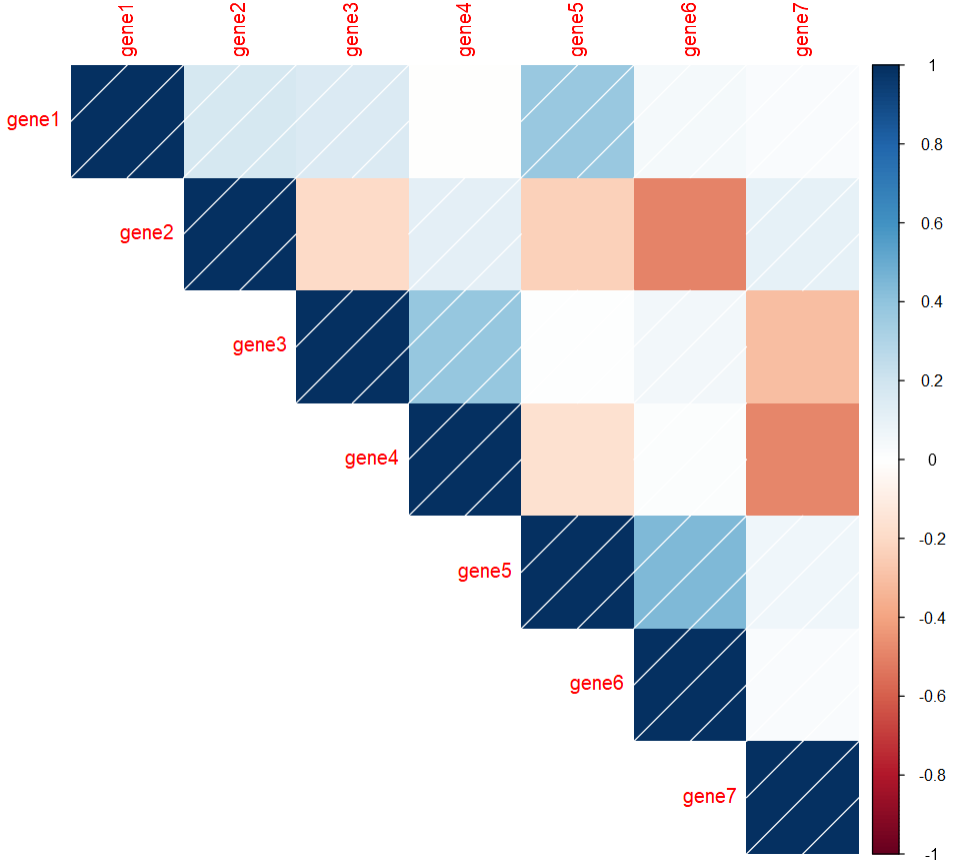
#对数字进行调整P8 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'number',#展示类型"full", "lower", "upper"type = 'upper',number.cex = 2, #数字大小number.font = 1, #字体形式number.digits = 2) #小数的保留结果
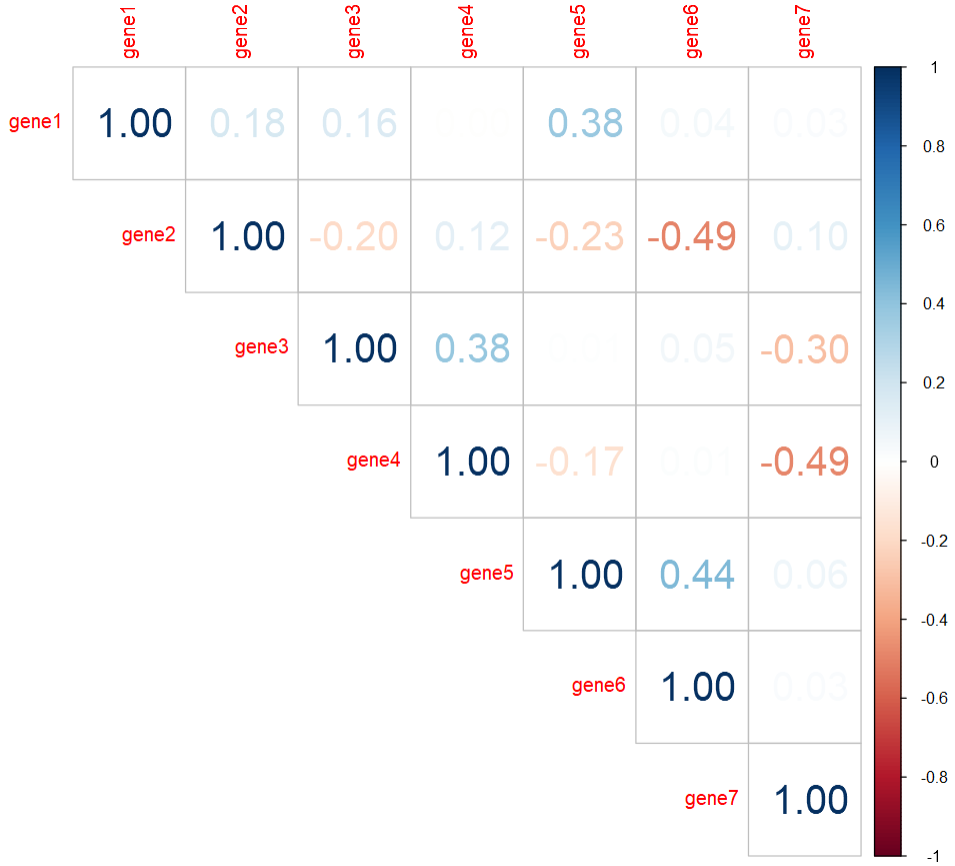
#拼图P9 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'number',#展示类型"full", "lower", "upper"type = 'lower',col = col, #改变颜色number.cex = 1, #数字大小number.font = 1, #字体形式#add= T, #添加另一个图#改变文本标签的位置'lt', 'ld', 'td', 'd' or 'n'. 'lt'tl.pos = 'd',tl.col = 'black',number.digits = 2 #小数的保留结果)P9 <- corrplot(cor, #简单绘图#改变形式"circle", "square", "ellipse", "number", "shade", "color", "pie"method = 'color',#展示类型"full", "lower", "upper"type = 'upper',col = col, #改变颜色,# bg = 'black' , #改变背景色title = 'test for corrplot',is.corr = T , #输入是否为相关矩阵,一般都是Tadd = T , #可通过改变type合并两种类型的图diag = F, #是否展示对角线结果#改变文本标签的位置'lt', 'ld', 'td', 'd' or 'n'. 'lt'tl.pos = 'd', #可用于两个图拼接时使用tl.cex = 0.8,tl.col = 'black', #文本大小和颜色cl.pos = 'n')

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |