新手上路,终于跑通了:利用edta注释基因组的重复序列(以拟南芥为例)
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
还记得小果之前分享了在Ubuntu系统中安装miniconda及edta吗,现在终于可以开始注释了,小果深刻阐释了想吃猪肉从养猪开始这一行为!
当天真的小果以为装好软件对代码复制粘贴(不是)就可以轻松注释时,悲剧就诞生了!因为作为一只菜果,她还不明白erro!erro!error!是能把人逼疯的。
废话不多说,我们还是进入正题吧!上期说要用到小鼠基因组,但是要练习的话还是选择基因组比较小的物种更快,所以小果换成了拟南芥。
1. 下载基因组数据并解压缩Arabidopsis thaliana genome assembly TAIR10.1 – NCBI – NLM (nih.gov)

在ncbi提供了3种方式,download可以直接下载到本地,datasets则要下载ncbi datasets进行安装,curl则需要已安装curl,比如说用curl安装则直接复制命令到终端就可以了。
在过程中需要用到genometools,我们用miniconda安装即可,不指定安装目录会默认安装到conda环境下的pkgs目录下。
conda install -c bioconda genometools-genometools
查看安装情况
conda list genometools
4. 开始注释!
conda activate EDTA
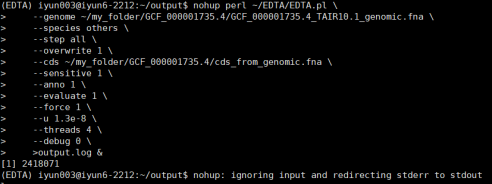
nohup perl ~/EDTA/EDTA.pl
–genome ~/my_folder/GCF_000001735.4/GCF_000001735.4_TAIR10.1_genomic.fna
–species others
–step all
–overwrite 1
–cds ~/my_folder/GCF_000001735.4/cds_from_genomic.fna
–sensitive 1
–anno 1
–evaluate 1
–force 1
–u 1.3e-8
–threads 4
–debug 0
>output.log &
为了更好理解,小果将代码的含义放在下面:
记得先激活EDTA环境哦!
nohup: 忽略SIGHUP信号,即使当前终端关闭也能继续运行。
–genome:指定待分析的基因组序列文件路径
–species:指定待分析基因组的物种信息,这里指定为“others”
–step:指定分析步骤,这里指定为“all”表示执行完整的分析流程
–overwrite:如果输出文件已经存在,是否覆盖,这里指定为“1”表示覆盖
–cds:指定包含CDS序列的文件路径,这个文件通常由基因组注释文件提供
–sensitive:是否使用较为敏感的TE识别算法,这里指定为“1”表示使用
–anno:是否使用已有的基因组注释信息进行分析,这里指定为“1”表示使用
–evaluate:是否对分析结果进行评估,这里指定为“1”表示使用
–force:是否强制执行,这里指定为“1”表示强制执行
–u:指定TE库中的最小不同种同族重复元件数量,这里指定为“1.3e-8”
–threads:指定使用的线程数量,这里指定为“4”
–debug:是否输出调试信息,这里指定为“0”表示不输出
edta.log:将标准输出重定向到名为“edta.log”的文件中
&:将进程放到后台运行。
接下来可以通过查看输出日志来查看进度
cat output.log
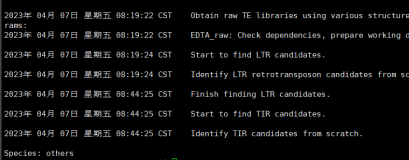
可以看到小果的进度目前是没有erro的~
小果在这一过程中犯了一些错误,给大家借鉴:
1.忘记激活EDTA 环境导致报错
2.没有安装genometools
3.路径错误
当然了,每个人遇到的问题可能不一样,在过程中可以根据自己的报错来解决问题。
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(处理大量数据的时候不用服务器用什么!快来联系我们使用吧!)

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓