xCell:基因表达数据的免疫浸润分析专家!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

-
hi大家,好久不见,小果又来啦! -
xCell是一种基于基因表达数据的免疫细胞浸润评估工具,可以识别出潜在的免疫细胞亚群并计算它们在组织中的相对丰度。它可以广泛应用于癌症、自身免疫性疾病和其他疾病的免疫浸润分析中。
今天小果就来教大家如何实战应用xCell进行基因表达数据的免疫浸润分析,那就和小果一起来看下吧!
数据预处理
下载xCell包并导入:
install.packages('devtools')devtools::install_github('dviraran/xCell')library(xCell)

xCell免疫浸润分析
导入基因表达矩阵,并通过xCellAnalysis分析表达矩阵,该函数用于预测细胞组分在给定样本中的相对丰度:
exprSet=read.csv("GSE57065_ExprMatrix.csv",header=T,row.names=1)xCell <- xCellAnalysis(exprSet) ##write.csv(xCell,"xCell_score.csv") #将计算后的结果写入表格文件中
先来一起看下分析后的数据集吧!
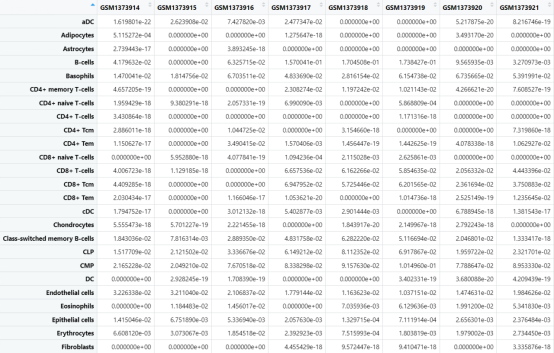

处理xCell分析后的结果数据集
接下来,我们可以根据样本的分组情况,提取出对应的细胞类型得分。
首先我们要读取 group.txt 的文件,这里包含了样本的分组信息,温馨提示,细胞表达样本数据包含两种类型哦!
我们可以读入这个文件,并提取出“septic_shock” 和 “healthy” 两组样本的得分:
group=read.delim("group.txt",header=T,stringsAsFactors = FALSE,sep="t")score=read.csv("xCell_score.csv",header=T,row.names=1,stringsAsFactors = FALSE)#取出结果文件中的前64个样本,并分别取出“septic_shock”,即带有疾病的样本群和“healthy”,即健康的样本群到对应的数据集中case_data=score[1:64,group$group=="septic_shock"]control_data=score[1:64,group$group=="healthy"]all(row.names(case_data)==row.names(control_data))normal=ncol(control_data)tumor=ncol(case_data)normal_data=as.data.frame(t(control_data))tumor_data=as.data.frame(t(case_data))#合并两种类型的结果文件rt_total = rbind(normal_data,tumor_data)cell_group_file=read.delim("cell_type.txt",header=T,stringsAsFactors = FALSE)cell_group=unique(cell_group_file$Subgroup)

整合样本分组信息与细胞分组信息
处理好分析后的数据后,我们可以通过进一步整合绘制所有结果数据集对应的热图,那就和小果一起来看一下怎样绘制吧!
在绘制热图之前,我们首先要整合数据、对所需数据进行筛选等操作,为后续的热图绘制工作做好准备工作,我们一起来看一下吧!
rt=t(rbind(normal_data,tumor_data)) #正常组和肿瘤组的基因表达矩阵按行合并,并进行转置,使样本在列上排列。cell_group_file=cell_group_file[order(cell_group_file$Subgroup),]rt=rt[cell_group_file$Cell.types,] #读入细胞类型信息文件cell_group_file,对其按照Subgroup这一列列进行排序all(row.names(rt)==cell_group_file$Cell.types)group1=c(rep("healthy",nrow(normal_data)), #合并健康组和肿瘤组样本rep("septic_shock",nrow(tumor_data)))names(group1)=colnames(rt)group1=as.data.frame(group1)type=cell_group_file[,2:3]row.names(type)=type$Cell.typestype$Cell.types=NULL
生成热图
做好上述准备工作后,我们可以导入pheatmap包并绘制热图,热图的具体绘制方法可以看小果之前发过的笔记哦,接下来和小果一起来看一下绘制过程吧!
library(pheatmap)pdf("Heatmap.pdf",height=8,width=12)pheatmap(rt,annotation_col =group1,annotation_row=type,annotation_colors =list(Type=c(healthy = "#42B540FF", septic_shock = "#925E9FFF")) ,color = colorRampPalette(c("green", "black", "red"))(50),cluster_cols =F,fontsize = 9,fontsize_row=8,fontsize_col=6,cluster_rows = F,show_colnames = F)dev.off()
好啦!现在你已经成功完成了热图的绘制,接下来我们看看绘制效果:

xCell对分析结果下游分析
同样地,我们也可以对xCell分析的结果进行下游分析并通过小提琴图对结果进行直观的分析,和小果一起来看一下怎样操作吧!
现在我们提取出了xCellAnalysis分析后的样本数据集并做了封装处理,现在我们可以针对我们的细胞群数据进行下游分析以及可视化处理的操作。
主要的流程为:根据不同的细胞子群对样本进行分类,遍历每个细胞子群,并计算每个子群的差异显著性,并对显著差异的细胞子群进行绘图展示和保存结果。那么就让我们一起来看下代码实现吧!
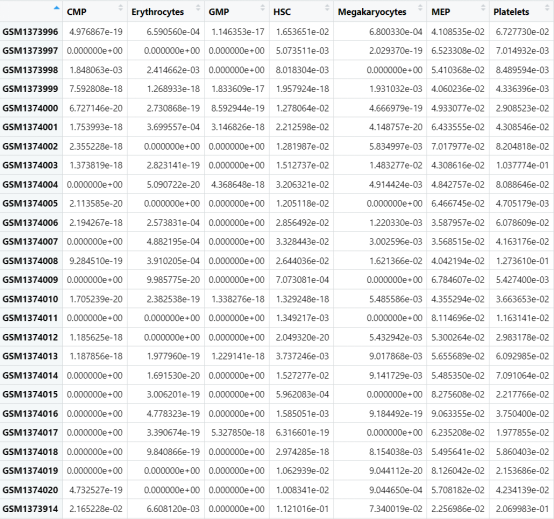
for (subgroup in cell_group){s=which( cell_group_file$Subgroup==subgroup) #获取当前细胞所属子群的indexcells=cell_group_file$Cell.types[s] #提取属于当前子群的所有细胞rt=rt_total[,cells] #提取属于当前子群的所有细胞类型的数据,并保存在 rt 中cell=c()p.value=c()for(i in 1:ncol(rt)){normalData=rt[1:normal,i]tumorData=rt[(normal+1):(normal+tumor),i]wilcoxtest=wilcox.test(normalData,tumorData,exact = F) #计算其差异的 p-value,并保存在p.value中,细胞类型名称保存在cell中p=round(wilcoxtest$p.value,3)p.value=c(p.value,p) #合并cell=c(cell,colnames(rt)[i])}sig=data.frame(cell,p.value)sig=sig[sig$p.value<0.05,] #选出p值小于0.05的显著细胞类型s=which(colnames(rt)%in%as.character(sig$cell))rt=rt[,s]all(colnames(rt)==as.character(sig$cell))
现在,小果给大家展示遍历到的其中一个细胞子群的最终计算结果数据,即我们提取出的要可视化的数据集,一起来看看吧!
通过小提琴图可视化下游分析的结果
现在,我们可以将最终的计算结果通过小提琴图展示出来,让我们来看看具体的代码实现吧!
library(vioplot)pdf(paste(subgroup,"Xcell_score.pdf",sep="_"),height=8,width=15) #设置可视化和保存的文件名称par(las=1,mar=c(10,6,3,3))x=c(1:ncol(rt))y=c(1:ncol(rt))#绘制空白散点图plot(x,y,xlim=c(0,(ncol(rt)-1)*3+2),ylim=c(min(rt),max(rt)+0.02),main="GSE57065",xlab="", ylab="Xcell score",pch=21,col="white",xaxt="n",cex.lab=1.3,cex.main=1.5)for(i in 1:ncol(rt)){normalData=rt[1:normal,i]tumorData=rt[(normal+1):(normal+tumor),i]wilcoxtest=wilcox.test(normalData,tumorData,exact = F)p=round(wilcoxtest$p.value,3)vioplot(normalData,at=3*(i-1),lty=1,add = T,col = '#42B540FF') #绘制正常数据的小提琴图vioplot(tumorData,at=3*(i-1)+1,lty=1,add = T,col = '#925E9FFF') #绘制肿瘤数据的小提琴图mx=max(c(normalData,tumorData))lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))text(x=3*(i-1)+1,y=mx+0.02,labels=ifelse(p<0.001,paste0("p<0.001"),paste0("p=",p)),cex = 0.8)}text(seq(1,((ncol(rt)-1)*3+1),3),-0.01,xpd = NA,labels=colnames(rt),cex = 1,srt = 45,pos=2,font=2)dev.off()write.csv(sig,paste(subgroup,"_sig_cell.csv",sep="")) #保存最终结果}
现在,你已经成功完成了xCell分析细胞到可视化的左右工作,现在小果给大家展示其中一个细胞子群的下游分析可视化结果!一起来看吧!
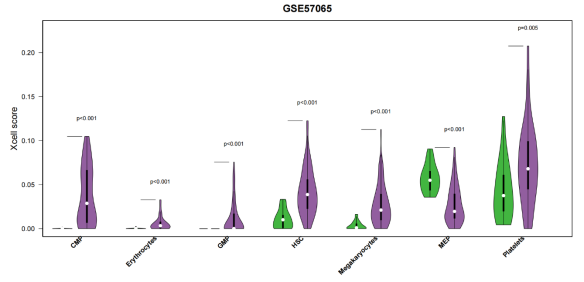
怎么样,你学会怎么使用xCell包了嘛? 更多学习资源请大家移步小果专属云生信平台搜索更多资源哦!
小果专属云生信平台:云生信 – 学生物信息学 (biocloudservice.com)
云生信平台也有免疫专版的学习模块哦,快来找到你想学习的专属模块吧!如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)

微信号 | 18502195490
知乎 | 生信果
点击“阅读原文”立刻拥有
↓↓↓