答应我,一定要看!基因差异表达——调控生物发育的神秘机制!使用BaySeq包带你探索基因差异表达的奥秘!!

公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240416
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("baySeq") # 在BiocManager环境下安装baySeq查看是否安装成功packageVersion("baySeq") # 查看baySeq版本

library(baySeq) # 载入baySeq包if(require("parallel")) cl <- makeCluster(4) else cl <- NULL # 定义一个cluster来实现并行处理,从而优化分析速度。data(simData) # 载入模拟数据集simData[1:10,] # 显示模拟数据前十行。

replicates <- c("simA", "simA", "simA", "simA", "simA", "simB", "simB", "simB", "simB", "simB") # 定义复制结构名称groups <- list(NDE = c(1,1,1,1,1,1,1,1,1,1), + DE = c(1,1,1,1,1,2,2,2,2,2)) # 分组并加上标签CD <- new("countData", data = simData, replicates = replicates, groups = groups)libsizes(CD) <- getLibsizes(CD)
plotMA.CD(CD, samplesA = "simA", samplesB = "simB", + col = c(rep("red", 100), rep("black", 900))) # 绘制MA图
CD@annotation <- data.frame(name = paste("count", 1:1000, sep = "_")) #添加注释信息CD <- getPriors.NB(CD, samplesize = 1000, estimation = "QL", cl = cl) # 获得负二项分布的先验分布CD <- getLikelihoods(CD, cl = cl, bootStraps = 3, verbose = FALSE) # 获得后验似然,估计差异表达计数的比例topCounts(CD, group = "DE") 
plotPosteriors(CD, group = "DE", col = c(rep("red", 100), rep("black", 900))) 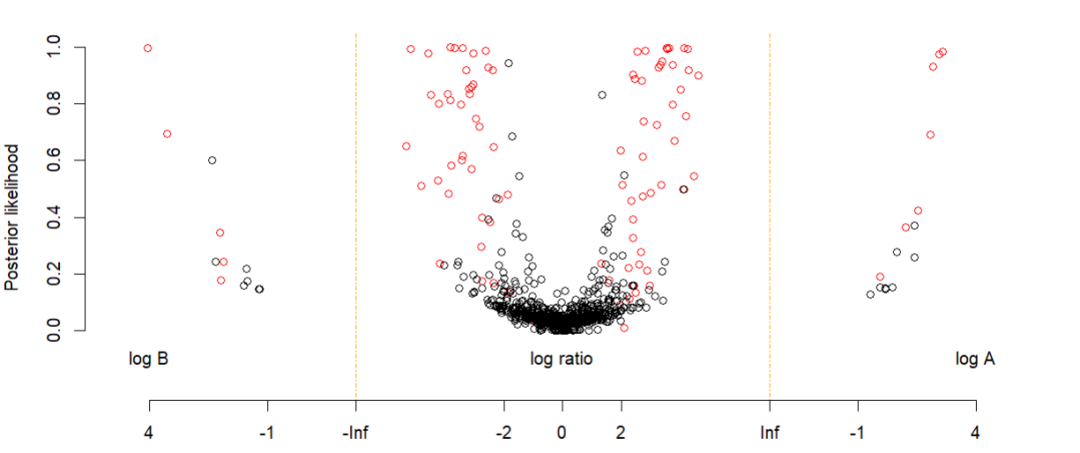
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果
往期回顾
|
01 |
|
02 |
|
03 |
|
04 |