基因差异性分析三大利器大揭秘,让你秒懂如何选择!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

基因数据的差异性分析是一种针对不同生物样本或不同处理条件下基因表达量变化的分析方法。它可以用来识别基因在两个或多个样本之间的表达差异,从而帮助我们了解基因在不同生物状态下的功能和调控机制。对于基因芯片的差异表达分析,由于其数据普遍被认为服从正态分布,因此常用的差异表达分析方法是在每个基因上应用t检验或方差分析。而高通量分析涉及到多个基因的同时检验,因此需要进行多重比较校正,以控制假阳性的发生。
基因数据的差异性分析通常包括以下步骤:
数据预处理:包括对原始数据进行质量控制、去除掉低质量的基因和样本、进行归一化等。
差异性分析:通过统计学方法(如t检验、方差分析、Wilcoxon检验等)或机器学习方法(如支持向量机、随机森林等)来比较不同样本之间的基因表达量,找出表达量显著差异的基因。差异性分析的结果可以用Volcano图或Heatmap等方式进行可视化展示,以便更好地理解差异基因的特点和表达模式。
功能富集分析:对差异基因进行功能富集分析,可以帮助我们了解这些基因在生物学过程、通路、功能和疾病中的作用和调控机制。功能富集分析方法包括基因本体论分析、富集分析、通路分析等。
结果解释和验证:基于差异性分析和功能富集分析的结果,可以进一步进行实验验证,以确定差异基因的生物学意义和功能,从而更好地理解基因在不同生物状态下的作用和调控机制。
DESeq2,EdgeR和limma是三种R语言中常用的差异表达分析工具包,可以用于分析RNA-seq或microarray等高通量数据的差异表达。
DESeq2:一般差异分析软件如DESeq2和edgeR的原理是比较组间差异和组内差异。所以说通常情况下,如果没有重复,则组内差异无法计算。DESeq2采用数据归一化和去除批次效应的方法,以消除样本之间的技术变异。它通过对样本中的基因计数数据进行负二项式模型拟合,来鉴定基因在不同样本之间的表达差异并进行差异基因的统计显著性检验。同时也可用于考虑生物学重复的影响,从而更准确地估计基因表达量的方差和标准误差。DESeq2使用Wald检验来鉴定差异基因,并使用Benjamini-Hochberg方法来进行多重检验校正。另外,DESeq2还提供了多种可视化方法,如MA图、Heatmap和PCA分析等。
##DESeq差异分析dds <- estimateSizeFactors(dds) 计算归一化系数sizeFactordds <- estimateDispersions(dds) 估计基因的离散程度dds <- nbinomWaldTest(dds) 统计检验,差异分析

EdgeR:适用于RNA-seq和microarray数据。使用负二项式模型来对基因表达数据进行建模,使用TMM(trimmed mean of M values)方法进行数据归一化。EdgeR使用exact test或quasi-likelihood F-test来进行差异分析。同时,它也使用Benjamini-Hochberg方法来进行多重检验校正。edgeR有一部分功能是依赖于limma包。

limma:目前在基因芯片的分析用的最多的就是limma。虽然它是针对芯片数据开发的,但也有limma-voom可以分析转录组数据。在处理RNA-Seq数据时,raw read count先被转成log2-counts-per-million (logCPM),然后对mean-variance关系建模。limma使用线性模型来分析microarray和RNA-seq数据。通过经验Bayes方法来调整基因表达值,以提高差异基因的检测能力,并使用FDR控制方法进行多重检验校正。limma也提供了许多可视化方法,如MA图、Heatmap和Volcano图等。
小果以limma为例,带大家实践一下吧~
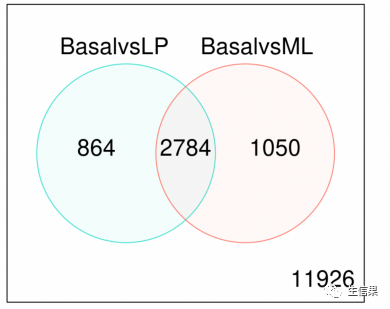
design <- model.matrix(~0+group+lane)colnames(design) <- gsub("group", "", colnames(design))contr.matrix <- makeContrasts(BasalvsLP = Basal-LP,BasalvsML = Basal - ML,LPvsML = LP - ML,levels = colnames(design))par(mfrow=c(1,2))v <- voom(x, design, plot=TRUE)vfit <- lmFit(v, design)vfit <- contrasts.fit(vfit, contrasts=contr.matrix)efit <- eBayes(vfit)summary(decideTests(efit))tfit <- treat(vfit, lfc=1)dt <- decideTests(tfit)summary(dt)de.common <- which(dt[,1]!=0 & dt[,2]!=0)length(de.common)vennDiagram(dt[,1:2], circle.col=c("turquoise", "salmon")#韦恩图展示了仅basal和LP(左)、仅basal和ML(右)的对比的DE基因数量,还有两#种对比中共同的DE基因数量(中)。在任何对比中均不差异表达的基因数量标于右下。
library(gplots)basal.vs.lp.topgenes <- basal.vs.lp$ENTREZID[1:100]i <- which(v$genes$ENTREZID %in% basal.vs.lp.topgenes)mycol <- colorpanel(1000,"blue","white","red")heatmap.2(lcpm[i,], scale="row",labRow=v$genes$SYMBOL[i], labCol=group,col=mycol, trace="none", density.info="none",margin=c(8,6), lhei=c(2,10), dendrogram="column"#在basal和LP的对比中前100个DE基因log-CPM值的热图。调整后,每个基因(每行)#的表达均值为0,并且标准差为1。给定基因相对高表达的样本被标记为红色,相对低表达 #的样本被标记为蓝色。浅色和白色代表中等表达水平的基因。样本和基因已通过分层聚类的 #方法重新排序。图中还显示有样本聚类的树状图。

以上就是基因差异性分析常用的R包~欢迎大家来和小果交流哦!
还有更多方便实用的小工具在云生信平台等着你哦~
http://www.biocloudservice.com/home.html
如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:(http://www.biocloudservice.com/home.html),其中也包括了通路表达分析(http://www.biocloudservice.com/313/313.php),单细胞的基因共表达分析(http://www.biocloudservice.com/906/906.php)等各种小工具哦~,有兴趣的小伙伴可以登录网站进行了解。
点击“阅读原文”立刻拥有
↓↓↓