超级侦探SingleR,揭露单细胞数据细胞类型的奥秘

嗨,果粉儿们!今天小果要和大家一起探索的是单细胞数据的世界,准确的说,我们要和一个超级侦探SingleR一起揭露这些细胞的奥秘。如果你曾经苦恼于如何给单细胞数据打上正确的标签,而非一堆无意义的数字;或者你也尝试过各种各样的细胞注释方法,却发现它们要么太复杂,要么太主观,无法满足你的需求。那么今天的内容一定能帮到你,跟上小果的步伐,我们出发啦。
 SingleR的前世今生
SingleR的前世今生
SingleR是一个用于对单细胞RNA测序数据进行细胞类型自动注释的R包,它于2019年由Aran等人发表在Nature Immunology上(https://www.nature.com/articles/s41590-018-0276-y )。SingleR的开发源于作者对单细胞数据注释方法的不满。作者认为,目前的单细胞数据注释方法大多依赖于人工选择的marker基因或者基于聚类的假设,这些方法都存在一定的偏差和局限性。因此,作者提出了这个基于参考数据集的无偏差的单细胞数据注释方法。
SingleR的核心思想是利用已知细胞类型的参考转录组数据集,通过计算每个单细胞与参考细胞之间的相似度,来推断每个单细胞的最佳匹配细胞类型。SingleR不需要用户提供任何先验信息或参数,也不需要用户进行任何聚类或降维操作,只需要输入单细胞数据和参考数据集,就可以得到每个单细胞的细胞类型标签。
Sounds amazing? 那么如何使用SingleR呢?其实很简单,只需要三步:
第一步:安装和加载SingleR包
#从Bioconductor安装if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("SingleR")#加载library(SingleR)
第二步:准备输入数据
SingleR需要两个输入数据:一是我们要注释的单细胞数据(称为test data),另一个是你要用来匹配的参考数据集(称为reference data)。test data应该是一个包含了基因表达量的矩阵或对象,其中每一行代表一个基因,每一列代表一个细胞。reference data应该是一个包含了纯细胞类型转录组数据和对应标签的矩阵或对象,其中每一行代表一个基因,每一列代表一个参考细胞。
##输入的参考数据集
如果你没有自己的参考数据集,不用担心,SingleR提供了7个内置的参考数据集供你选择。目前有以下几个选项:
Human Primary Cell Atlas (HPCA):包含了32种人类主要细胞类型的转录组数据集,来源于Immunological Genome Project (ImmGen)和Human Cell Atlas (HCA)
Blueprint Encode Data:包含了713种人类造血细胞类型的转录组数据集,来源于Blueprint和ENCODE项目
Monaco Immune Data:包含了28种人类免疫细胞类型的转录组数据集,来源于Monaco等人的研究
Novershtern Hematopoietic Data:包含了38种人类造血细胞类型的转录组数据集,来源于Novershtern等人的研究
Mouse RNAseq Data:包含了38种小鼠细胞类型的转录组数据集,来源于ImmGen项目
你可以根据你的数据的物种和细胞类型来选择合适的参考数据集。这里我们选用的是人类肾脏数据,所以选择Human Primary Cell Atlas作为参考数据集
#可以用celldex包的内置函数直接加载参考数据集:ref.hpca <- HumanPrimaryCellAtlasData()#或者导入之前下载好的参考数据集ref.hpca <- get(load("hpca.Rdata"))##需要注释的单细胞数据这里我们选择了一套包含4000多个细胞的人类肾脏样本作为输入数据:https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSM5225906&format=file&file=GSM5225906%5Fs200929N8%2Eexpression%5Fmatrix%2Etxt%2Egz# 导入表达矩阵。data <- read.table(file="GSM5225906_s200929N8.expression_matrix.txt.gz")# 导入所需的库library(Seurat)library(SingleR)library(celldex)#常规的数据预处理,创建一个Seurat对象(scRNA)。scRNA <- CreateSeuratObject(counts = data, min.cells = 3,min.features = 200, project = "kidney")scRNA <- SCTransform(scRNA)scRNA <- RunPCA(scRNA, verbose = F)scRNA <- RunUMAP(scRNA, dims = 1:15)scRNA <- FindNeighbors(scRNA, dims = 1:15) %>% FindClusters(resolution = 0.6)#为节约时间,我们选取前500个细胞sce <- scRNA[,1:500]#获取标准化矩阵expdata <- GetAssayData(sce, slot="data")
第三步:进行singleR注释
#进行注释,注意SingleR自带参考集的标签是分别是main,ont和fine,fine和ont表示更细分的标签,这里我们用main的结果就可以了。sce.hpca <- SingleR(test = expdata, ref = ref.hpca, labels = ref.hpca$label.main)#设置细胞类型的标签sce@meta.data$celltype <-sce.hpca$labels#可视化DimPlot(sce, group.by = c("seurat_clusters", "celltype"),reduction = "umap")
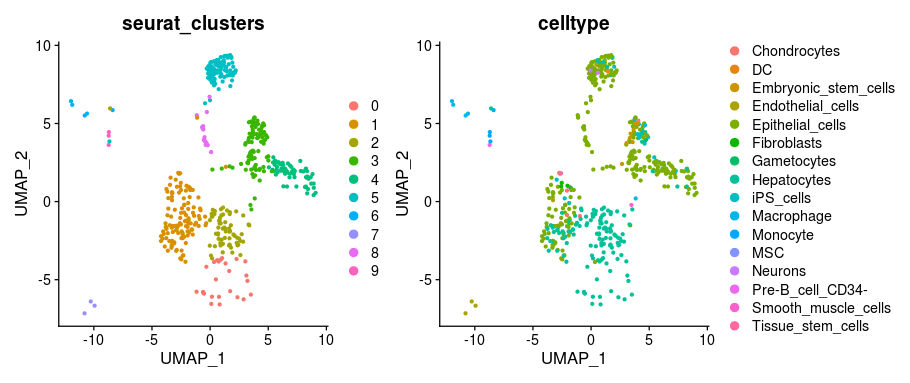
如果想要更换参考数据集只需一键导入
ref.be <- BlueprintEncodeData()并重复第三步即可
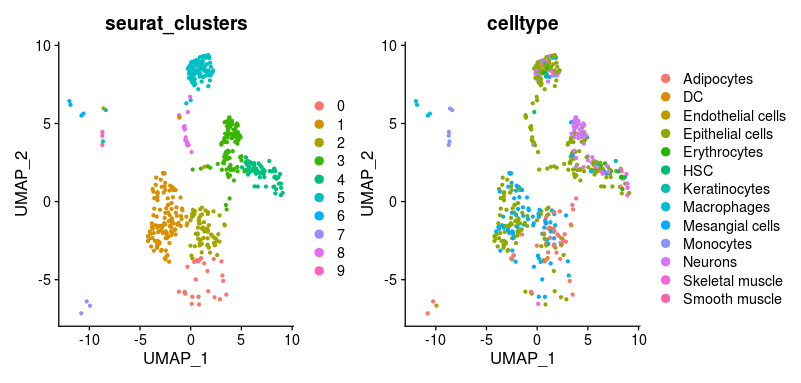
可以发现更换参考集之后,注释的细胞类型发生了变化。
当然这只是最基础的细胞注释手段,是否准确还需要进一步评估,想要学习更高阶的细胞注释,请持续关注小果的后期分享。
我们今天的探索就要结束了。希望你已经了解了如何使用这个超级侦探SingleR来揭露你的单细胞数据中的细胞类型的奥秘。快试试看吧,也可以成为一个超级的单细胞数据科学家!
往期推荐

