【crosshap】小果教你GWAS的局部单倍型可视化
本期小果给大家分享一个用于GWAS(全基因组关联分析)的局部单倍型可视化的R包,它2023年8月1日发表在Bioinformatics期刊上的,又是一个新鲜出炉的好包!
无论你是本科生还是研究生,无论你专攻湿试验还是生信,这篇推文将为你提供深入了解这一令人激动的新技术的机会。让我们一起探索交叉单体型聚类(crosshap)如何改变我们对遗传学的认识,以及它在未来的研究中可能发挥的作用。请看小果下面的分享!
前言
GWAS技术以其出色的性能,成功地利用了大规模的基因变异数据集,以识别可能导致特定表型的候选区域。然而,传统的GWAS和精细定位方法无法深入了解包含并受到导致性状变异的遗传变异影响的局部连锁基因组结构。
小果将带领大家探讨一项令人激动的新技术 – crosshap,它为我们提供了一种更深入了解基因组内部复杂结构的方法。传统的GWAS可能已经为我们揭示了一些关于基因和表型之间的联系,但crosshap带来了一种全新的视角,让我们能够更详细地探究基因之间的关系,以及它们如何影响我们的遗传特征。
它通过基于变异连锁配置文件的密度分析,实现对感兴趣的局部基因组区域中的单体型结构的稳健聚类。随后,crosshap还配备了可视化工具,用于选择最佳的聚类参数(ɛ),然后生成一个直观的图表,提供了连接变异、单体型组合、表型和元数据特征之间复杂关系的概览。
安装及用到的数据
我们将分析的数据来自美国农业部核心收集的大豆数据(Bayer等人,2021年)。小果接下来将使用crosshap(Marsh等人,2022年)复现种子蛋白QTL、cqProt-003的局部单倍型分析。您可以从crosshap示例数据的figshare仓库中下载这些数据。
公众号后台回复“111″,领取代码,代码编号:231016
figshare.com/articles/dataset/fin_b51_173kb_only_vcf/22331167install.packages("devtools")devtools::install_github("jacobimarsh/crosshap")library(crosshap)
输入数据
我们下载数据后,保存到对应的路径,读取即可。
#Read example data into R and standardize using helper functionsvcf <- read_vcf('path/to/fin_b51_173kb_only.vcf')#VCF is provided as additional argument to help assign column and row namesLD <- read_LD('path/to/plink.ld', vcf = vcf)pheno <- read_pheno('path/to/sprot_phen.txt')metadata <- read_metadata('path/to/namepopfile.txt')
局部单倍型
局部单体型涉及通过在感兴趣区域内的位点上共享的单体型(等位基因组合)来分类个体。在CrossHap中,单体型并不是由单个位点来定义的,而是由标记组(Marker Groups)的等位基因组合来定义的。标记组是紧密连锁的SNP(单核苷酸多态性)的集群,通常在整个人群中作为一个单元遗传。
用于捕获标记组的聚类算法是DBSCAN(Ester等人,1996年),在CrossHap中有两个主要参数:MGmin和epsilon。
MGmin(minPts)是一个簇中必须具有的最小链接SNP数,才能将其分类为标记组,而不是背景噪声。在下面的示例中(173kb区域中的2041个SNP),MGmin等于30。
#Add minimum Marker Group SNP countMGmin <- 30
Epsilon
Epsilon大致对应着簇密度,当epsilon增加时,通常会包括更多的SNP在更多的标记组中。在这个阶段,值得提供一系列epsilon值,以便稍后选择最佳值。由于这个数据集包含大量的SNP(2041位点),所以我们将测试的epsilon范围将会相对较低(0.2 – 1)。
#Add list of epsilon values to run haplotyping onepsilon <- c(0.2,0.4,0.6,0.8,1)
现在输入文件和参数已经设置好啦,是时候进行单倍型分析了!
#Run the haplotyping at all provided epsilon valuesHapObject <- run_haplotyping(vcf = vcf,LD = LD,pheno = pheno,metadata = metadata,epsilon = epsilon,MGmin = MGmin)
如果一切顺利,现在已经保存了一个单体型对象(HapObject),其中包含了所有五个提供的epsilon值的单体型结果。这个单体型对象将需要用来构建CrossHap的可视化(以及选择的epsilon值以进行可视化!)
优化聚类分辨率
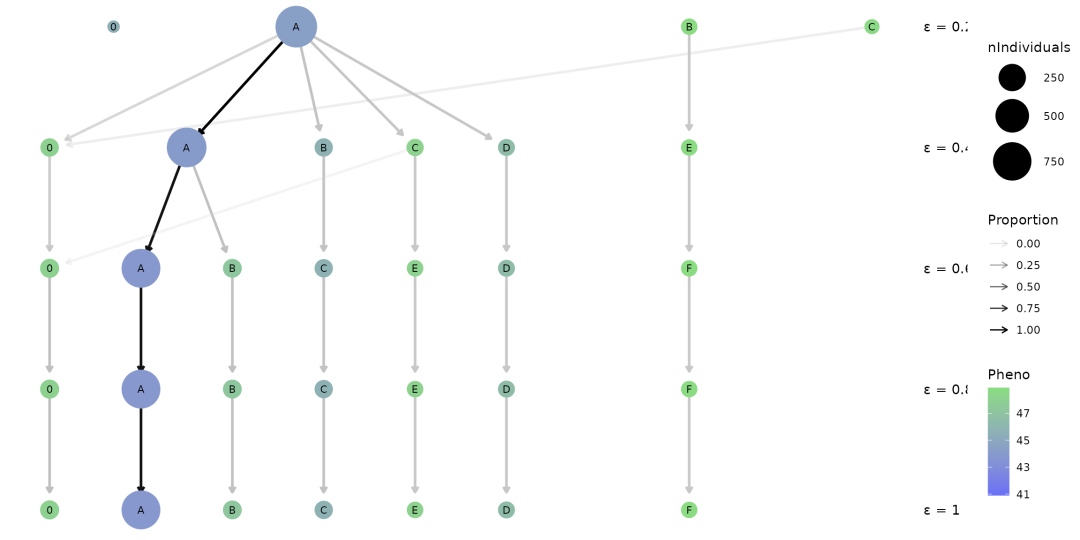
Epsilon是DBSCAN中一个非常难以优化的参数,这是用于将SNP聚类成CrossHap中的标记组,以进行单体型分析。幸运的是,crosshap有一个clustree(Zappia等人,2018年)工具,它可以用于总结不同epsilon值下创建的单体型对象之间的差异,让我们可以选择最佳的epsilon值。
第一个聚类树选项提供了在不同epsilon值下解析的单倍型组的变化摘要。
#HapObject created by `run_haplotyping`#Add type = 'hap' to ensure it summarizes haplotypes rather than Marker Groupshap_clustree <- clustree_viz(HapObject,type = 'hap')hap_clustree
可视化局部单倍型
现在我们已经将结果(单倍型对象)准备好了,并且已经为我们的数据集选择了最佳的epsilon分辨率,是时候将我们的结果完整地可视化了,这样我们就可以开始发现变化模式了!
在十字形可视化中,中心矩阵显示了定义每个单倍型的Marker Group等位基因的组合,它提供了一个网格,围绕着每个Marker Group和单倍型组合的特征。单倍型的特征和分配给它们的个体是垂直可视化的,在顶部和底部的图中有特征。标记组的特征和分配给它们的snp水平可视化,特征在左边和右边的图上。
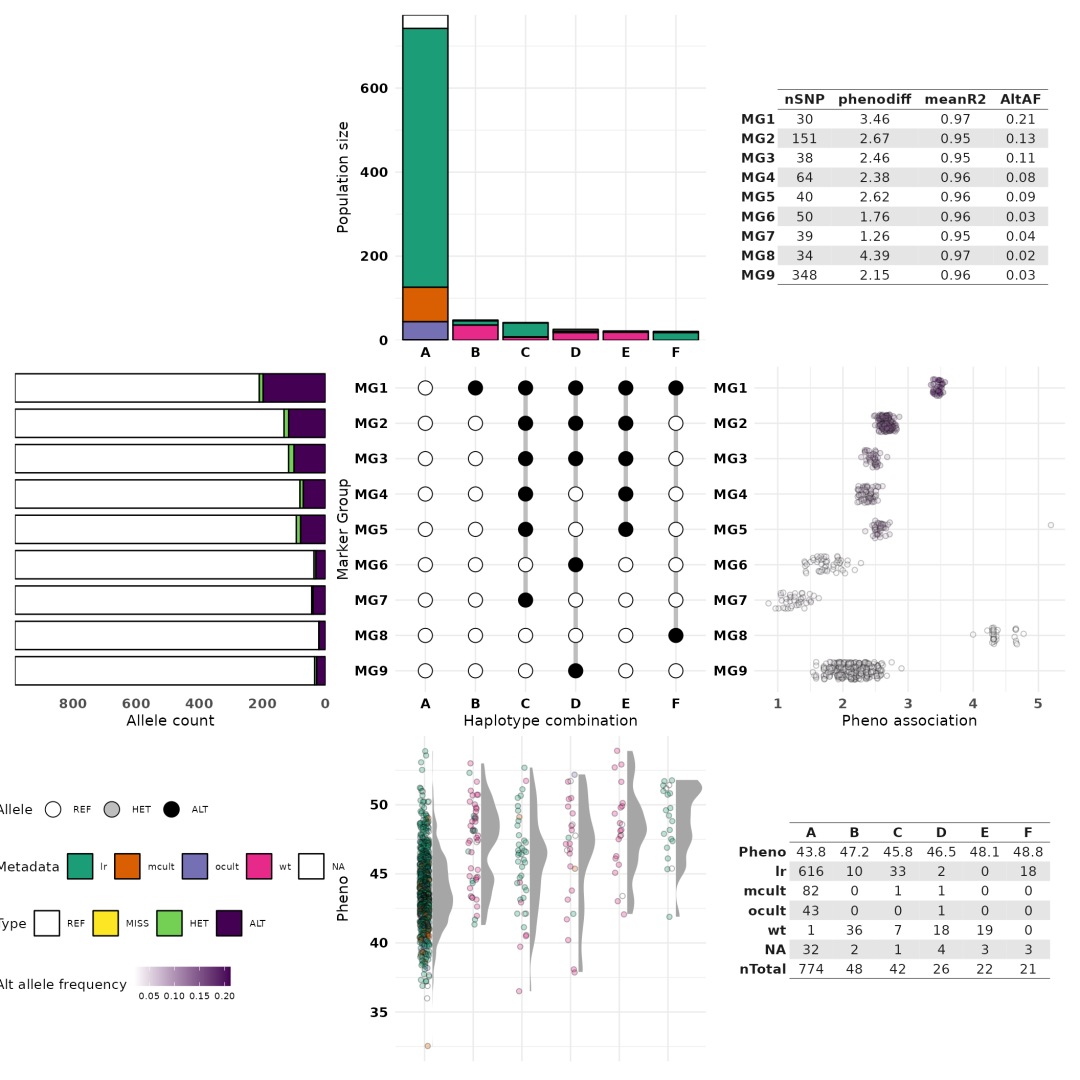
这些图都是什么意思?
-
在中央矩阵上方,显示了每种单体型组合的个体频率,还有按我们提供的元数据变量(驯化程度水平)进行的分解。
-
下方,绘制了每个个体的表型得分(种子蛋白质含量百分比),每个点代表一个个体,根据单体型分组,并根据元数据变量进行着色,就像顶部的条形图一样。
-
左侧,是每个标记组(MG)的等位基因频率信息摘要。这显示了每个标记组内所有SNP中次要等位基因、缺失等位基因、杂合子和参考等位基因的频率。
-
右侧,绘制了每个SNP的表型关联(每个点代表一个SNP),根据标记组进行分组。”表型关联” 实际上是指对于给定的SNP,拥有替代等位基因和参考等位基因的个体之间的表型得分差异。
-
右下角的表格总结了单体型信息,的表格总结了标记组信息。所有的图注都显示在左下角。
结语
小果今天给大家分享了这个用于GWAS的局部单倍型可视化的R包,大家之后在做GWAS的时候不要忘记这个工具哦!
参考文献
[1] Marsh JI, Petereit J, Johnston BA, Bayer PE, Tay Fernandez CG, Al-Mamun HA, Batley J, Edwards D. crosshap: R package for local haplotype visualization for trait association analysis. Bioinformatics. 2023 Aug 1;39(8):btad518. doi: 10.1093/bioinformatics/btad518. PMID: 37607004; PMCID: PMC10471896.
[2] https://jacobimarsh.github.io/crosshap/articles/Getting_started.html
往期推荐


