rrvgo: 从零开始,快速上手简化富集分析与可视化的利器
{ 点击蓝字,关注我们 }
小师妹今天将向您介绍如何使用R软件包rrvgo来简化Gene Ontology(GO)富集分析结果中的冗余信息。rrvgo可以根据基因之间的语义相似度,将相似的GO富集结果合并为更高级别的节点,从而提供更简洁和易理解的富集结果。
1)什么是GO富集分析?
GO富集分析是一种用于研究基因集合功能的常用分析方法。它通过将基因与功能注释之间的关联进行统计,识别在给定基因集合中显著富集的功能项(GO节点)。GO节点包括三个主要分支:生物过程(BP)、分子功能(MF)和细胞组分(CC)。富集分析的结果通常是一个包含富集节点、基因数、调整的p值等信息的表格。
2)问题:GO富集结果中的冗余信息
在进行GO富集分析时,可能会遇到一个问题,即富集结果中存在大量的冗余信息。这是因为不同的基因集合可能会显著富集相同的功能节点,导致结果中出现重复的GO节点。冗余信息给结果的解释和可视化带来困难,同时增加了分析的复杂性。
3)解决方案:使用rrvgo简化GO富集结果
rrvgo可以帮助我们解决上述的冗余问题。它通过计算基因之间的语义相似度,将相似的GO富集结果合并为更高级别的节点,从而减少结果中的冗余信息。简化后的富集结果更具可读性,便于结果的解释和可视化。
现在,让我们一起学习如何使用rrvgo包来简化GO富集结果。
一
安装和加载rrvgo包
首先,我们需要安装和加载rrvgo包,这样才能在R环境中使用相关的函数和功能。
在R控制台中执行以下命令:
代码:
if (!require("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("rrvgo")library("rrvgo")
二
准备GO富集结果数据
在使用rrvgo之前,我们需要准备GO富集分析的结果数据。结果数据通常以表格形式存储,包含有关富集节点、基因数、p值等信息。
确保您有一个包含GO富集结果的数据框或文件。数据框应至少包含以下列:
-
GO_ID:富集节点的唯一标识符。
-
Description:富集节点的描述或名称。
-
GeneCount:富集节点中的基因数。
-
P-value:富集节点的p值或调整的p值。
例如,以下是一个示例GO富集结果数据框:
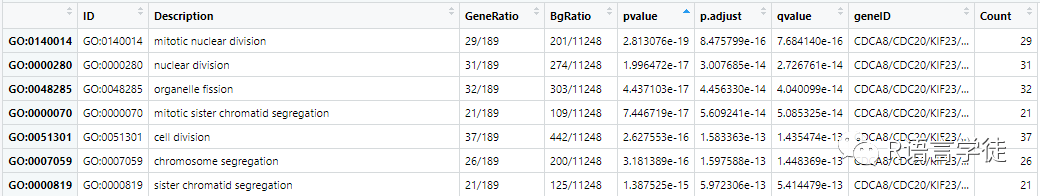
图1 示例GO富集结果
如果您的数据存储在文件中,您可以使用适当的函数(例如read.table()或read.csv())将其加载到R环境中。
代码:
go_analysis <- read.delim(system.file("extdata/example.txt", package="rrvgo"))三
计算基因之间的语义相似度矩阵
Rrvgo提供calculateSimMatrix函数计算语义相似度,该函数是使用GOSemSim包来实现的。
代码:
simMatrix <- calculateSimMatrix(go_analysis$ID, orgdb="org.Hs.eg.db", ont="BP", method="Rel")四
简化GO富集结果
现在,我们可以使用rrvgo来简化GO富集结果,使用reduceSimMatrix函数,将GO富集结果数据和基因之间的语义相似度矩阵作为输入。
代码:
scores <- setNames(-log10(go_analysis$qvalue), go_analysis$ID)reducedTerms <- reduceSimMatrix(simMatrix, scores, threshold=0.7, orgdb="org.Hs.eg.db")
scores是基于GO富集结果的得分向量,它表示每个GO节点的重要性或显著性。执行reduceSimMatrix()函数后,将得到一个简化后的语义相似度矩阵reducedTerms。这个矩阵将只包含较为显著且不冗余的GO节点。
通过调整threshold参数的值,可以控制简化的程度。较高的阈值将导致更严格的简化,可能会删除较多的节点,而较低的阈值将保留更多的节点。在选择阈值时,需要根据具体问题的需求和分析目标进行权衡
五
可视化结果
rrvgo 提供了几种绘制和解释结果的方法。
1)相似度矩阵热图
代码:
heatmapPlot(simMatrix, reducedTerms, annotateParent=TRUE, annotationLabel="parentTerm", fontsize=6)这将使用simMatrix作为输入数据,绘制一个具有颜色编码的热图,显示不同节点之间的相似度。您可以根据需要进行自定义,调整热图的颜色、标签、注释等参数。
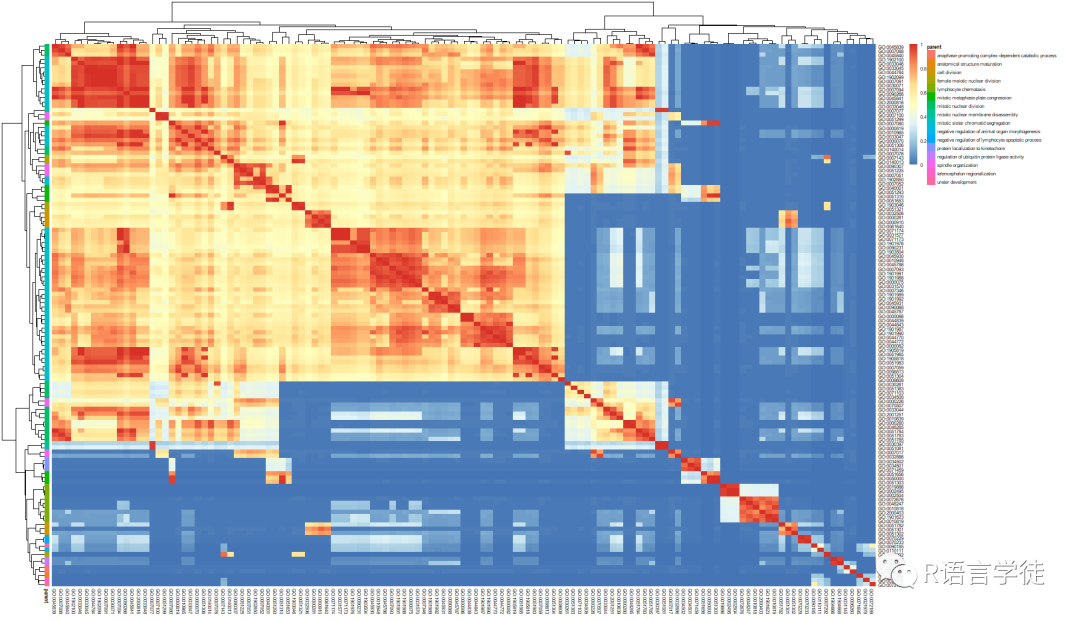
图2 相似性度热图
2)描述组和terms之间距离的散点图
scatterPlot函数用于绘制terms之间的散点图,点于点的距离显示它们之间的相似性,点的大小代表根据p值计算的分数,或者在没有分数的情况下,GO terms包含的基因数量。
代码:
## 散点图scatterPlot(simMatrix, reducedTerms)

图3 散点图
3)树状图
rrvgo包中的treemapPlot()函数用于绘制基于GO terms的树状图。树状图可以帮助可视化GO terms之间的层次结构和关系,terms所占的空间大小与分数成正比。
代码:
##树状图treemapPlot(reducedTerms)
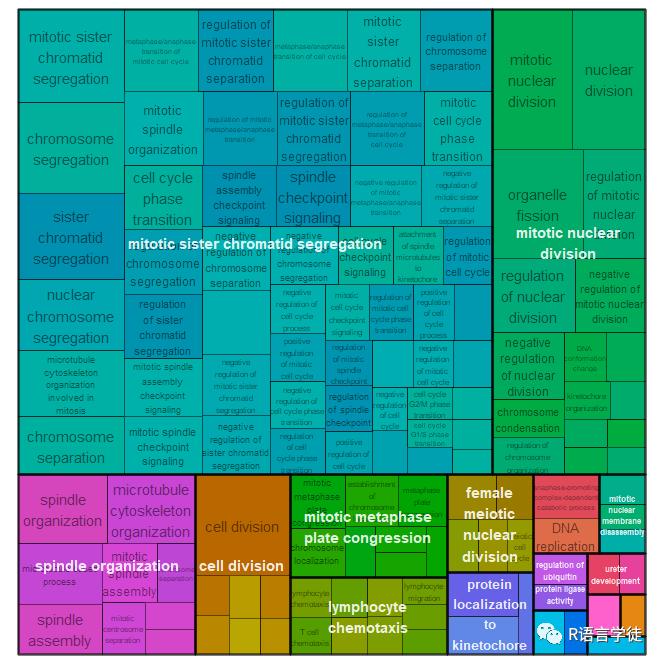
图4 树状图
4)词云图
rrvgo包中的wordcloudPlot()函数用于绘制基于GO terms的词云图。词云图可以帮助可视化GO terms的频率或权重,并根据其在数据集中的重要性调整词的大小。
代码:
## 词云图wordcloudPlot(reducedTerms, min.freq=1, colors="black")
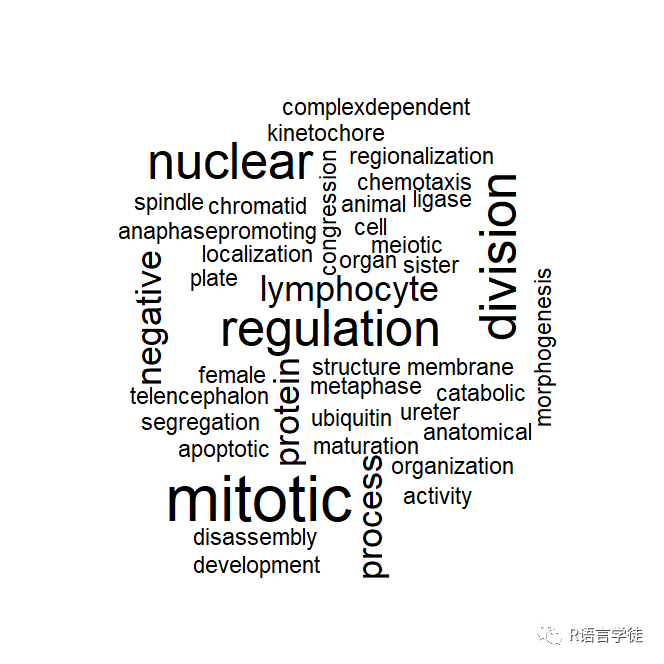
图5 词云图
根据您的需求和喜好,您可以使用不同的图表类型来创建更具吸引力和有意义的图表。
以上就是小师妹给大家介绍的使用rrvgo包简化GO富集结果的基本步骤。通过简化结果,您可以获得更清晰、更易解释的富集结果,从而更好地理解基因集合的功能特征。本教程提供了一个基本的指导,供新手入门。根据您的具体数据和分析需求,您可能需要进一步探索rrvgo包的功能和参数,以优化分析结果。希望这篇教程对您有所帮助!更多学习资源请大家移步小师妹专属云生信平台(云生信 – 学生物信息学 (biocloudservice.com))搜索更多资源哦!
E
N
D
