TCGA原始表达谱数据如何处理?一个R包帮你搞定!
点击蓝字 关注我们
生物信息学最常用的数据库有哪些?大海哥认为,不管有谁,肯定不能少了TCGA(The Cancer Genome Atlas),赞同的小伙伴点个赞^-^
TCGA(The Cancer Genome Atlas)是一个旨在帮助深入了解多种癌症类型的项目,通过收集、分析和分享来自多种癌症患者的大规模基因组数据,以更好地了解癌症的分子机制、诊断标志物和治疗方法。该项目使用了多种高通量技术,其中我们最常使用的是来源于RNA测序(RNASeq)技术的基因表达谱数据,但一般给出的测序数据是包含编码和非编码RNA的。对于不同课题组经常会有不同的研究倾向,需要从数据中将所有非编码RNA提取或剔除出去。其实可以通过Excel等方式来操作,但那样其实很麻烦,今天大海哥带大家用R语言来实现!
开始之前,大家先了解一下什么是Ensembl ID、Entrez ID和Gene symbol?
Gene ID 也称Entrez ID/EntrezGene ID ,是 NCBI 使用的能够对众多数据库进行联合搜索的搜索引擎, 其对不同的 Gene 进行了编号, 每个 gene 的编号就是 entrez gene id,就是一串数字,比如:TP53 的Gene ID是:7157。因为entrez ID相对稳定, 所以也被其他数据库, 如 KEGG 等采用。不同物种的同一个基因的Gene ID是不同的。
Gene Symbol ,是HGNC数据库为基因提供的官方名称,HGNC是人类基因命名委员会(HUGO Gene Nomenclature Committee);人类基因组命名委员会。有专门的数据库:https://www.genenames.org/。主要是按基因的功能起的名字,字母一般为英文全称的缩写,由大写字母和数字组成,如TP53基因的Official Symbol就是由HGNC所提供。
Ensembl ID,是在Ensembl数据库中对基因的命名,常见的物种前缀:“ENS“表示Homo sapiens (Human),”ENSMUS“表示Mus musculus (Mouse),”ENSDAR“表示Danio rerio (Zebrafish);而常见的序列类型用G、P、T、分别表示gene、protein和transcript。TCGA下载的数据一般就是用的Ensembl ID。
那么了解完基本概念后,我们就使用今天的主角R包:BioMart来开始操作吧!
# 加载所需R包suppressPackageStartupMessages({library(biomaRt)library(tidyverse)})#使用biomaRt创建转换表biomart_table <- getBM(attributes = c("external_gene_name","ensembl_gene_id","entrezgene_id","gene_biotype"),mart = useMart("ensembl", dataset = "hsapiens_gene_ensembl"))#看看长啥样
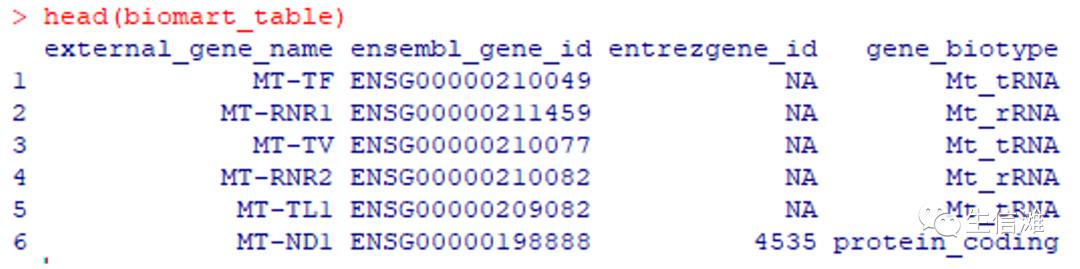
#########对于不同物种,需要修改dataset,可以使用如下代码查看:##mart = useMart('ensembl')##listDatasets(mart)

#有很多物种哦!自由挑选~# 使用NA代替空值biomart_table[biomart_table == ""] <- NA#保留几个关键信息biomart_table <- biomart_table %>%dplyr::select("Symbol" = external_gene_name,"Ensembl" = ensembl_gene_id,"Entrez" = entrezgene_id,"Biotype" = gene_biotype) %>%arrange(Symbol, Ensembl, Entrez, Biotype)#保存结果注释文件save(biomart_table, " biomart_table_homo.RData")#注释文件经过处理,最后包含以下这几列
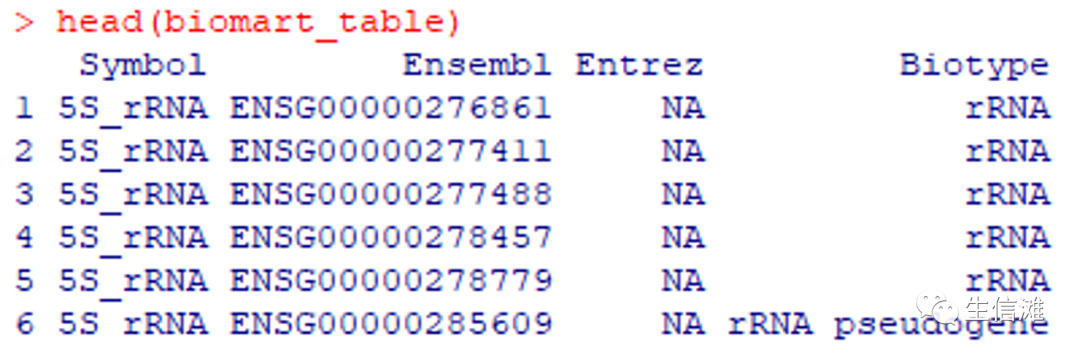
至此,就可以得到注释文件,接下来的任务就是匹配,可以使用Excel,方便的话还是用R:
load(“biomart_table_homo.RData”)
results <- read.delim(“diffexpr-results.txt”) #这是大海哥的数据哦!只用要求数据第一列为Ensembl ID就可以了,其他没有要求
results2 <- merge(results,biomart_table[c(2,1,4)],by = “Ensembl”)
results2 <- merge(biomart_table[c(2,1,4)],by = “Ensembl”,results)
write.csv(results2,”results.csv”)
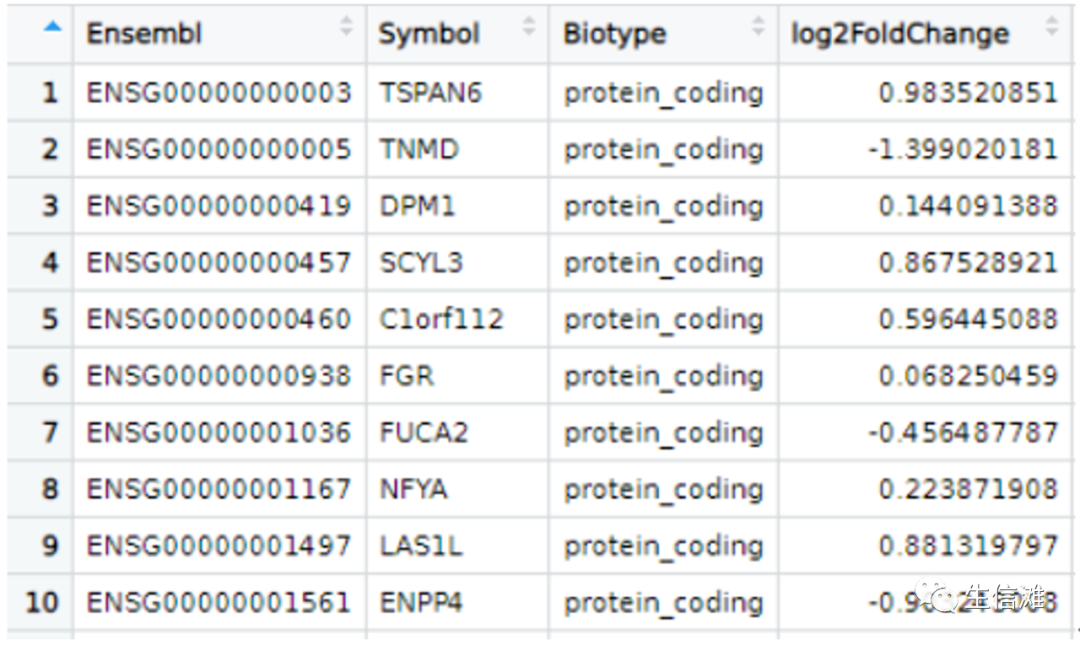
#然后可以看到获得了对应的Symbol列#此外,还可以根据这个包进行人鼠同源基因名字转化,使用getLDS函数,它是biomaRt查询的主要功能,连接两个数据集,并从这些链接的biomaRt数据集检索信息。在Ensembl中,这转化为同源映射。#使用亚洲镜像human <- useEnsembl('ensembl',dataset = "hsapiens_gene_ensembl", mirror = "asia")mouse <- useEnsembl('ensembl',dataset = "mmusculus_gene_ensembl", mirror = "asia")#比如我有一系列小鼠基因:mouse.genes <- c("Psme2b","Zfp661","Tsc22d1","Prox2","Eml3","Creb1")#将其映射到人的基因上MtoH <- getLDS(attributes = "mgi_symbol", # 要转换符号的属性,这里基因名(第3步是基因名)filters = "mgi_symbol", #参数过滤mart = mouse, #需要转换的基因名的种属来源,也就是第2步的mousevalues = mouse.genes, #要转换的基因集attributesL = "hgnc_symbol", #要同源转换的目标属性,这里还是转为基因名,也可加其他martL = human, #要同源转换的目标种属,也就是第2步的humanuniqueRows = TRUE)#但是目前好像这个getLDS功能有点异常,所以大海哥又想了个新办法#大海哥使用Orthology.eg.db包创建了一个比对函数,也可以实现人鼠基因转换mapfun <- function(mousegenes){gns <- mapIds(org.Mm.eg.db, mousegenes, "ENTREZID", "SYMBOL")mapped <- select(Orthology.eg.db, gns, "Homo_sapiens","Mus_musculus")naind <- is.na(mapped$Homo_sapiens)hsymb <- mapIds(org.Hs.eg.db, as.character(mapped$Homo_sapiens[!naind]), "SYMBOL", "ENTREZID")out <- data.frame(Mouse_symbol = mousegenes, mapped, Human_symbol = NA)out$Human_symbol[!naind] <- hsymbout}#需要导入以下三个包哦!没下载的小伙伴使用BiocManager::install(“”)可以下载library(Orthology.eg.db)library(org.Mm.eg.db)library(org.Hs.eg.db)mapfun(mouse.genes)
可以看到输出结果还是很顺利的,有一个基因转换没有结果,可能是数据库里没有这个基因,大家也可以自行去一些线上数据库检索一下哦!


点击“阅读原文”进入网址