两分钟带你了解R语言包“limma”,带你快速进行基因表达数据分析
点击蓝字 关注我们
Limma(Linear Models for Microarray Data)是一个在基因表达数据分析中广泛应用的R语言包。它提供了一套强大的统计方法和工具,用于差异表达分析和其他类型的基因表达数据分析。
下面是Limma包的一些主要功能和特点:
1. 线性模型:Limma包基于线性模型方法进行差异表达分析。它使用线性模型来建立基因表达数据和样本条件之间的关系,考虑到可能的批次效应和其他混杂因素。
2. 基因表达差异分析:Limma包提供了一套用于差异表达分析的统计方法。它可以识别在不同样本条件下基因表达的显著差异,并为每个基因计算调整的p值和表达差异度量。
3. 贝叶斯方法:Limma包使用贝叶斯方法来估计基因的差异表达,并为每个基因提供一个调整的p值。这种方法在小样本情况下表现出色,可以更好地处理低表达基因和噪声数据。
4. 批次效应调整:Limma包可以校正实验中的批次效应,提高差异分析的准确性。它提供了多种方法来检测和调整批次效应,包括ComBat等。
5. 多组差异分析:Limma包支持多组比较的差异分析。你可以在多个样本条件之间进行比较,并获得每个基因的差异表达结果和统计显著性。
6. 多重检验校正:Limma包提供了多种多重检验校正方法,如Benjamini-Hochberg方法和q值方法,以控制差异分析中的假阳性发现。
7. 可视化工具:Limma包包含了一些用于可视化差异表达结果的函数。你可以绘制基因表达的散点图、MA图、热图等,以直观地展示基因的差异表达模式。
8. 整合其他数据:Limma包还支持将基因表达数据与其他类型的数据(如蛋白质互作网络、生物通路注释等)进行整合分析,从而获得更全面的生物学解释。
要使用Limma包,可以在R中使用以下命令进行安装和加载:
> install.packages(“Limma”) # 安装Limma包> library(Limma) # 加载Limma包
示例:
这个示例使用了公开可获取的GSE1297数据集,该数据集包含急性淋巴细胞白血病(ALL)和急性髓系白血病(AML)患者的基因表达数据。以下是使用R语言包limma进行差异表达分析的示例。
# Load the required packages> library(limma)> library(GEOquery)> library(R.utils)# Download and load the gene expression data from GEO> gse <- getGEO("GSE1297", GSEMatrix = TRUE)> expression_data <- as.data.frame(exprs(gse[[1]]))> colnames(expression_data) <- pData(gse[[1]])$title# Define the experimental design> sample_info <- pData(gse[[1]])> design_matrix <- model.matrix(~ 0 + sample_info$characteristics_ch1.2)# Perform differential expression analysis using limma> normalized_data <- normalizeBetweenArrays(expression_data)> fit <- lmFit(normalized_data, design_matrix)> fit <- eBayes(fit)> results <- topTable(fit, coef = 1, adjust.method = "fdr", number = Inf)
# Volcano plot> volcano_plot <- limma::plotMA(fit, coef = 1, highlight = 10, names = NULL)

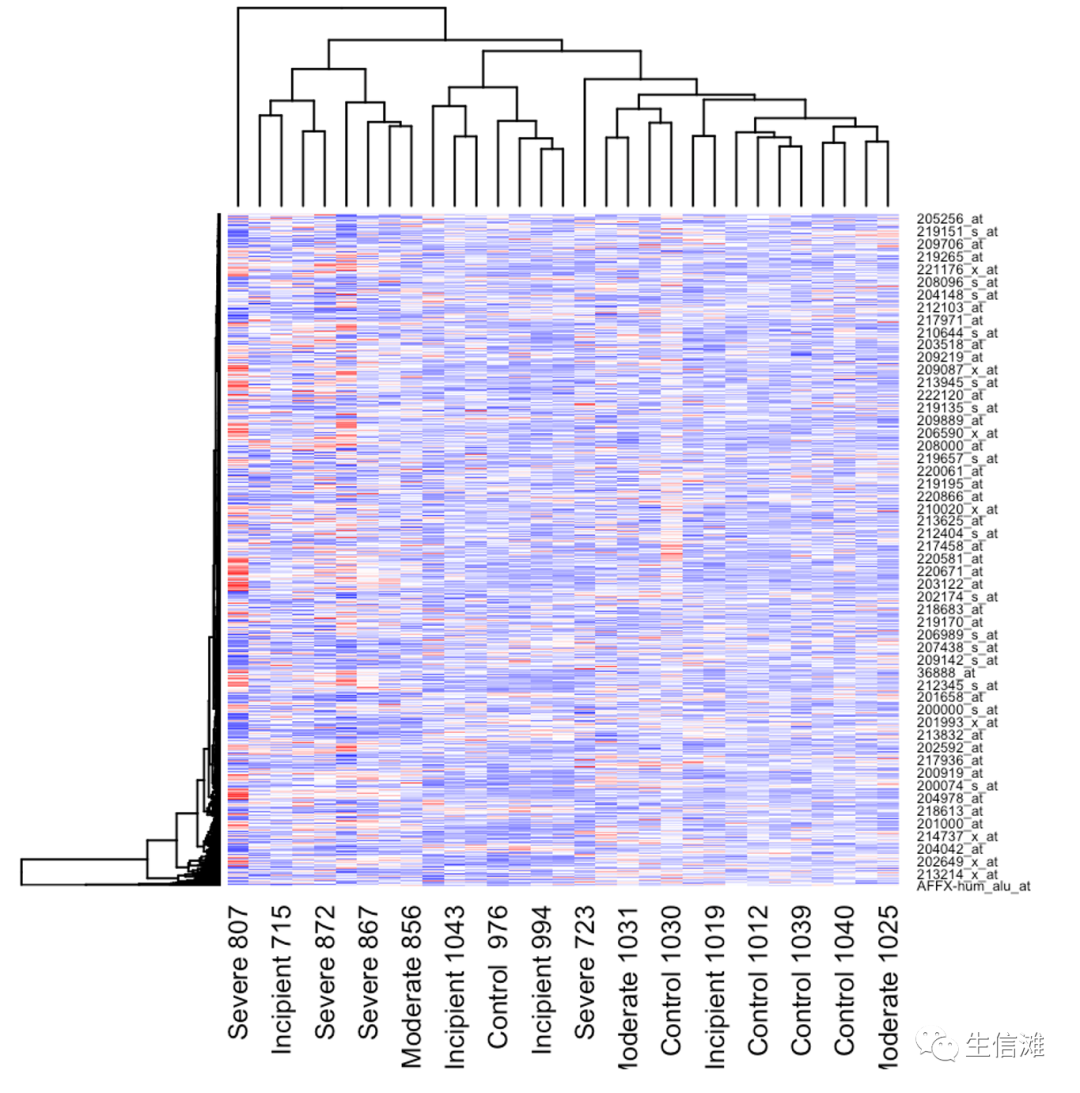
# Select differentially expressed genes> DE_genes <- rownames(results)[results$adj.P.Val < 0.05 & abs(results$logFC) > 1]> heatmap_data <- normalized_data[DE_genes, ]# Plot heatmap of differentially expressed genes> heatmap(heatmap_data, scale = "row", col = colorRampPalette(c("blue", "white", "red"))(100))
在这个示例中,我们首先加载了必要的包,包括limma和GEOquery。然后,我们使用getGEO函数从GSE1297数据集中下载基因表达数据。我们通过提取表达值和样本信息来对数据进行预处理。
接下来,我们使用样本信息定义实验设计。使用limma中的lmFit和eBayes函数执行差异表达分析。我们根据调整后的p值和折叠变化提取显著差异表达的基因。
最后,我们使用plotMA函数创建Volcano图来可视化差异表达结果。我们还使用heatmap函数生成差异表达基因的热图。
以上就是对R语言包Limma的简单介绍啦,Limma包在基因表达数据分析中被广泛应用,并在生物医学研究、生物信息学和系统生物学等领域发挥了重要作用。它具有灵活性、可靠性和可扩展性,适用于各种规模和类型的基因表达数据分析。


点击“阅读原文”进入网址