想用机器学习方法做预后分析?还不快来试试这个!
点击蓝字 关注我们
如果说什么技术的出现颠覆了传统生信圈,那大海哥觉得一定是机器学习,机器学习的出现给生物信息学带来了许多新的可能性和机会。例如在基因组学方面,通过机器学习可以在基因组学、生物信息数据挖掘、疾病诊断和治疗、药物发现以及系统生物学等方面都发挥出了巨大的促进作用,不仅提供了处理大规模生物数据的强大工具,还为我们研究人员们提供了新的研究思路和方法。
今天大海哥就来推荐一个简单好用且效率高的算法——CatBoost。咋一看名字,有些小伙伴可能不太了解,大家都只听过Gradient Boosting Machine(GBM)算法,CatBoost是什么算法,其实啊,CatBoost和GBM都属于梯度提升算法,都属于集成学习方法。只不过,CatBoost相比传统GBM,它具有一些优势和独特之处,CatBoost能够自动处理分类特征,无需额外的预处理(如独热编码)。它可以在训练过程中利用特征统计信息来将分类特征转换为数值特征,从而更好地利用这些信息。并且其引入了一种自动调参技术,可以通过设定一些全局参数来自动选择较为合适的学习率和树的数量等超参数,简化了模型调优的过程。它还能够处理缺失值,无需额外的处理方式。在决策树的分裂过程中,它会自动处理缺失值,并学习缺失值的模式。CatBoost采用了一种基于对称叶子分裂的算法,以减少过拟合风险。此外,它还通过组合随机排列技术引入了随机性,从而提高模型的泛化能力。关键的是它支持GPU运算哦!
说了那么多,这算法那么好,该怎么运用到生信数据上呢?别着急,大海哥准备了一个临床数据分析案例来演示这个算法。让我们开始吧!
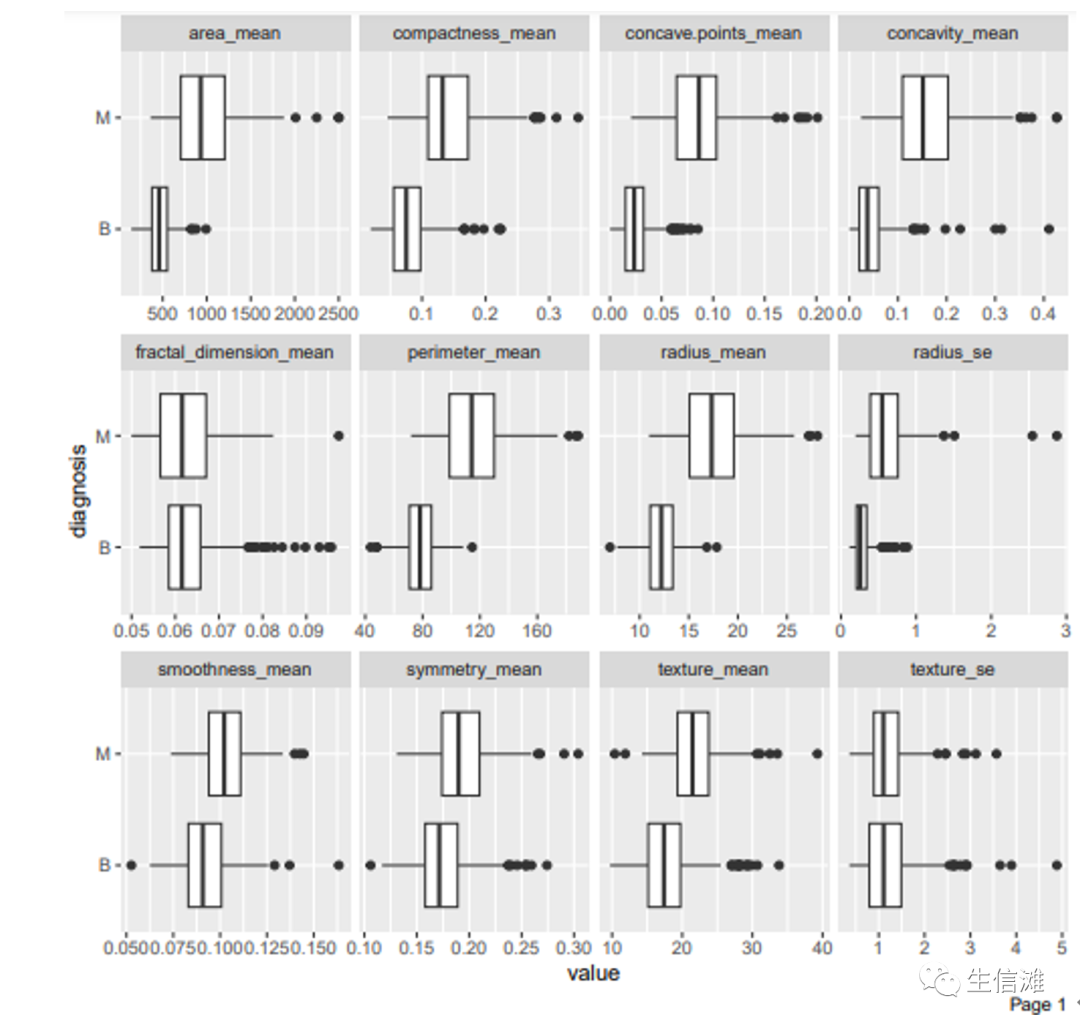
本次分析数据集一共包含32个变量,目标变量为病人的乳腺组织诊断结果,分别对应M(恶性)和B(良性)。特征变量包含病人的临床特征,例如对应肿瘤特征,包含大小,半径变化等病人临床信息的平均值、标准误差和最差值或最大值等共31个特征变量。
代码部分
#首先导入需要的R包library(tidyverse)library(caret)library(gbm)library(DataExplorer)library(pROC)#读取数据df_bc = read.csv("data.csv",check.names = F)[,1:32]#把数据里的ID去掉,同时对目标变量进行因子化df_bc01 = mutate(df_bc,diagnosis = as.factor(diagnosis))[,-1]#查看一下数据的列名colnames(df_bc01)

#看一下数据是否有缺失值plot_intro(df_bc01)

#画一个箱型图看看各个特征的数据分布情况plot_boxplot(df_bc01, by = "diagnosis")
#划分一下训练集和测试集set.seed(123)indice = createDataPartition(df_bc01$diagnosis, p = .75, list = F)train = df_bc01[ indice,]test = df_bc01[-indice,]#构建catboost模型set.seed(3456)fit_control_carboost <- trainControl(method = "repeatedcv",number = 5,repeats = 5,classProbs = TRUE)grid_carboost<-expand.grid(.iterations = 140, .rsm = 0.8, .border_count = 25, .depth = 1, .learning_rate = 0.1, .l2_leaf_reg = 1e-06)train_carboost<-train#开始训练catboost_01 <- train(x= train_carboost[ ,2:31],as.factor(make.names(train_carboost[,c("diagnosis")])),method = catboost.caret,logging_level = 'Silent',tuneGrid = grid_carboost, trControl = fit_control_carboost)#预测一下测试集previsioni_catboost_01 = predict(catboost_01, test[,-1])#计算一下混淆矩阵吧,看看精确度如何confusionMatrix(previsioni_catboost_01, as.factor(test$diagnosis))

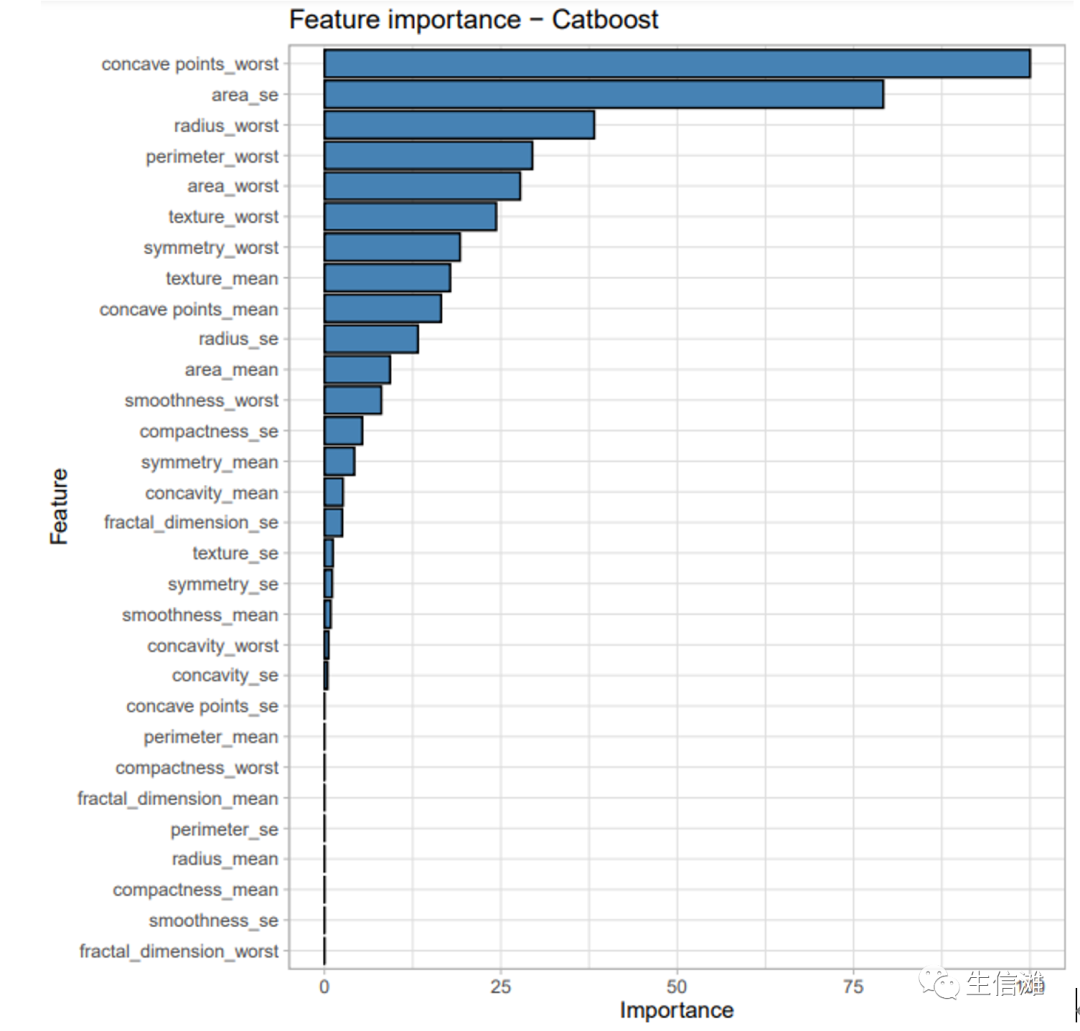
#结果相当不错哦!ACC值达到了0.9718,还可以看看不同的特征的重要性library(ggplot2)ggplot(varImp(catboost_01)) +geom_bar(stat = 'identity', fill = 'steelblue', color = 'black') +theme_light() +ggtitle('Feature importance - Catboost')
#最后画一个ROC曲线来评价一下ggroc(list(GBM=roc(as.numeric(test$diagnosis), as.numeric(previsioni_gbm_01)),Catboost=roc(as.numeric(test$diagnosis), as.numeric(previsioni_catboost_01))))+ggtitle("Comparing ROC Curves")

#可以看到结果很不错,代码的复杂度是不是也不高,同时不同于GBM的一点就是我们不需要设置决策树的数量,CatBoost会自行帮你选择最合适的决策树数量,十分便捷!
分析到这里就结束了,可以看到最后的CatBoost模型预测乳腺癌是否为恶性的准确率达到了97.18,并且concave points_worst和area_se是模型最为重要的两个参数。
点击“阅读原文”进入网址