差异分析魔法师:DESeq2与基因的魔幻冒险!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

还在为如何入门生信而彷徨吗?还在为Linux系统而感到无助吗?快关注小果的微信公众号,小果手把手带你入门生信,没有门槛、没有难度,跟着效果走,啥问题都没有。
hello,今天小果教大家如何使用DESeq2包对counts表达矩阵进行差异基因分析,并对分析结果绘制火山图实现差异基因分析的可视化,如果你感兴趣的话就和小果一起看下去吧!
首先小果先来简单介绍一下这个包:
DESeq2是一个R语言中常用的差异表达分析包,用于分析高通量测序数据中的差异表达基因。它是Bioconductor项目的一部分,提供了一套统计方法和功能,用于检测基因在不同条件下的表达差异,并找出具有生物学意义的差异表达基因。DESeq2包的设计目标是针对RNA-seq数据进行差异表达分析,并且能够处理具有低复制数、高变异性和有偏差的数据。它采用了负二项分布模型和广义线性模型(GLM)的方法,通过考虑样本之间的技术和生物学变异,进行准确的差异表达分析。
DESeq2包提供了一系列功能,包括:
1.数据导入:支持从不同的数据源导入RNA-seq数据,包括原始计数数据和已经进行了基因注释的数据。
2.数据预处理:包括数据规范化、去除低表达基因、去除批次效应等。
3.差异表达分析:使用负二项分布模型和GLM进行统计分析,计算差异表达基因的显著性。
4.多组实验设计:支持多组条件下的差异表达分析,包括两组比较、多组比较和时间序列分析。
5.基因注释和功能富集分析:支持将差异表达基因进行注释,并进行功能富集分析,以了解差异表达基因的生物学功能。
6.可视化:DESeq2包集成了ggplot2和其他可视化工具,可以方便地绘制差异表达基因的图表,如火山图、MA图等。
1、R包与数据加载
首先,设置工作目录为数据文件所在的路径,并加载DESeq2和其他所需的R包,并导入我们准备好的counts表达矩阵:
setwd("/media/desk16/iyun007/kegg")#BiocManager::install("DESeq2")library(DESeq2)data1 <- read.csv("Gene Symbol_matrix.csv",header = T, row.names = 1)
注意,我们导入的数据基本格式如下,同学们要提供符合要求的数据集哦!
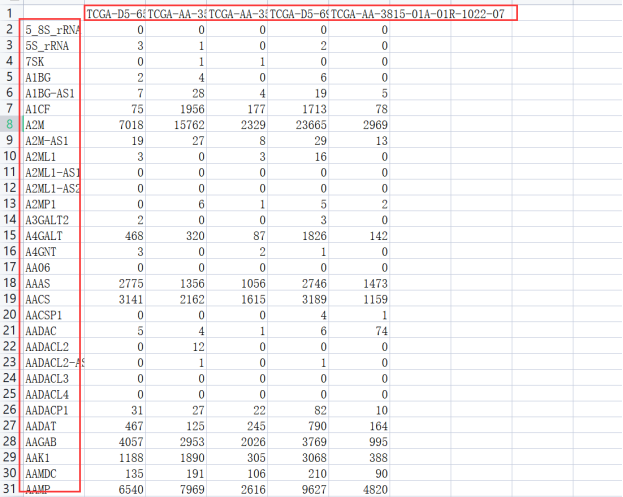
2、数据预处理
接下来,我们要构建分组矩阵,即根据数据框中样本的命名规则,提取出癌症样本和正常样本的列名,并将它们分别存储在tumor和normal向量中:
#构建分组矩阵tumor <- colnames(data1)[as.integer(substr(colnames(data1),14,15)) < 10] #data1表达矩阵中的所有癌症样本(8个)tumornormal <- colnames(data1)[as.integer(substr(colnames(data1),14,15)) >= 10] #data1表达矩阵中的所有正常样本(2个)normaltumor_sample <- data1[,tumor] #癌症样本的表达矩阵View(tumor_sample)tumor_samplencol(tumor_sample)normal_sample <- data1[,normal] #正常样本的表达矩阵View(normal_sample)ncol(normal_sample)data1 <- cbind(tumor_sample,normal_sample) #合并癌症和正常样本矩阵col_index <- which(colnames(matrix) == "normal_sample")colnames(matrix)[col_index] <- normal#colnames(data1)[5] <- "TCGA.AA.3525.11A.01R.A32Z.07"View(data1)group_list <- c(rep('tumor',ncol(tumor_sample)),rep('normal',1)) #根据自己的数据集设置tumor和normal数据的分组数condition = factor(group_list)coldata <- data.frame(row.names = colnames(data1), condition) #coldata是分组矩阵View(coldata)
最后合并好的数据如下:

分组信息如下:
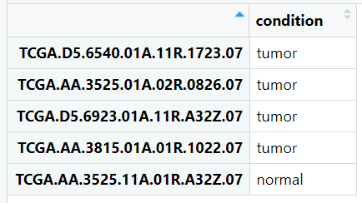
3、差异分析
接下来,我们可以:
1.创建DESeq2数据对象:使用DESeqDataSetFromMatrix()函数创建DESeq2所需的数据对象,将合并后的数据框和分组矩阵作为参数传递给该函数。
2.进行差异分析:使用DESeq()函数对DESeq2数据对象进行差异分析。
3.保存差异表达基因结果:将差异表达基因结果保存为CSV文件。
#构建dds对象,构建差异基因分析所需的数据格式dds <- DESeqDataSetFromMatrix(countData = data1, colData = coldata, design = ~ condition)#差异分析dds <- DESeq(dds)#提取结果result <- as.data.frame(results(dds)) # results()从DESeq分析中提取出结果表DGE <-subset(result,padj < 0.05 & (log2FoldChange > 2 | log2FoldChange < -2)) #这里我们提取出2倍差异表达、校正p值<0.05的显著差异表达基因DGE <- DGE[order(DGE$log2FoldChange),]write.csv(DGE,'DGE.csv',row.names = TRUE)
我们一起来看下分析后的结果吧!

4、绘制火山图
接下来,我们可以通过火山图可视化我们的分析结果,具体操作如下:
这里小果使用ggplot()函数创建绘图对象,并设置数据源为result数据框。然后使用geom_point()函数绘制散点图,其中x轴为log2FoldChange,y轴为-log10(padj),并根据差异表达的方向和显著性进行颜色分类。通过调整绘图的各种参数,如标题、坐标轴标签和图例,可以使火山图更具可读性和美观性哦。
## 5.提取结果result <- as.data.frame(results(dds)) # results()从DESeq分析中提取出结果表result <- na.omit(result)result1 <- result[c(2,6)]## 6.火山图result1$change = ifelse(result1$padj < 0.05 & abs(result1$log2FoldChange) >= 2, ifelse(result1$log2FoldChange> 2 ,'Up','Down'),'Stable')library('ggplot2')ggplot(result1, aes(x = log2FoldChange, y = -log10(padj),colour=change)) +geom_point(alpha=0.3, size=3.5) +scale_color_manual(values=c("#546de5","#d2dae2","#ff4757"))+labs(title='Volcano plot',x="log2(fold change)",y="-log10 (padj)")+ # 坐标轴# 坐标轴和图标题title="Volcano plot",theme_bw()+ #去除背景色#xlim(-20, 20)+ #设置坐标轴范围#ylim(0,40)+# 辅助线geom_vline(xintercept=c(-2,2),lty=3,col="black",lwd=0.8) +geom_hline(yintercept = -log10(0.05),lty=3,col="black",lwd=0.8) +theme(panel.grid = element_blank())+ #去除网格线theme(plot.title = element_text(hjust = 0.5,size=24),legend.position="bottom",legend.title = element_blank(),legend.text=element_text(size=18),legend.key.size = unit(1, 'cm'),legend.background = element_rect(fill="gray90", linetype="solid",colour ="gray"),axis.title.x =element_text(size=18),axis.title.y=element_text(size=18),axis.text=element_text(size=14,face = "bold"))ggsave("De_ana.png",width=8,height=6)# 保存为png格式
我们一起来看下最后的结果图吧!
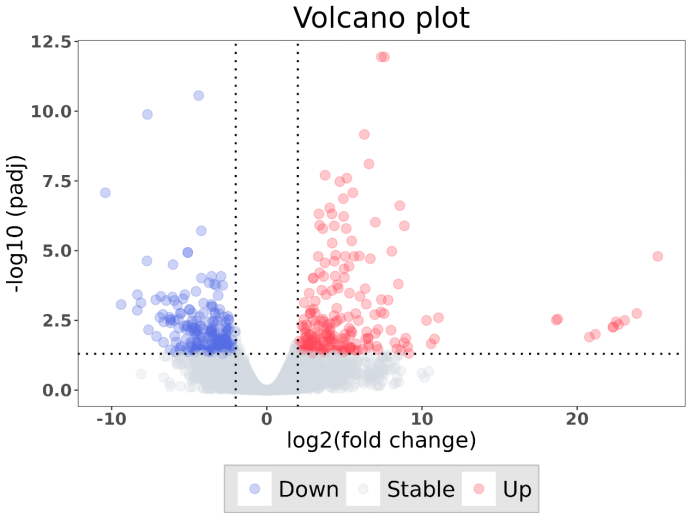
怎么样,你学会怎么使用DESeq2包来进行差异分析了嘛?是不是很简单!更多学习干活要继续关注小果哦!
这里推荐一下小果新开发的零代码云生信分析工具平台包含超多零代码小工具,上传数据一键出图,感兴趣的小伙伴欢迎了解~
网址:http://www.biocloudservice.com/home.html
今天小果的分享就到这里,欢迎大家和小果一起讨论学习,下期再见哦!
小果友情推荐
好用又免费的工具安利


