【ggpicrust2】小果教你宏基因组功能预测下游分析
 宏基因组研究可以说是生信的一大热门领域,今天小果给大家带来关于宏基因组功能预测下游分析的包——ggpicrust2(2023年8月新鲜出炉)。
宏基因组研究可以说是生信的一大热门领域,今天小果给大家带来关于宏基因组功能预测下游分析的包——ggpicrust2(2023年8月新鲜出炉)。
预测宏基因组功能有很多工具,包括PICRUSt2(Douglas et al. 2020)、Tax4Fun2 (Wemheuer et al. 2020)、MicFunPred (Mongad et al. 2021)、PICRUSt (Langille et al. 2013)……在这些工具里面,PICRUSt2可以说是皇冠上的明珠,它刊登在nature biotechnology上(小果的梦刊),已经帮助数以千计的研究者预测宏基因组功能。
图1 PICRUSt2(Douglas et al. 2020)
工具虽好,但学术界对如何推断并可视化PICRUSt2产生的功能丰度输出仍然没有达成共识。由于使用差异丰度(DA)方法确定群体之间功能和通路的统计显著差异是分析中的关键步骤,所以得选个好的DA方法。
PICRUSt2官方最初推荐使用STAMP)(Parks et al. 2014)作为分析和可视化的首选软件。然而,STAMP自2015年以来一直没有更新,所以它没办法整合最新的数据,而且它在macOS上安装很困难。为了解决上述问题,R语言包ggpicrust来了,是的它来了,它可以进行广泛的擦一风度(DA)分析,而且提供出版级可视化。
ggpicrust2是2023年8月1日发表在Bioinformatics期刊上,github已有46颗星,小果专门给大家分享新鲜出炉的包哦!

图2 ggpicrust2(Chen et al. 2023)
下面小果就带大家一起看看如何使用ggpicrust2吧!
公众号后台回复“111”领取本篇代码、基因集或示例数据等文件;
文件编号:231206
果粉福利:生信人必备神器——服务器
平时生信分析学习中有要的小伙伴可以联系小果租赁,粉丝福利都是市场超低价格,赶快找小果领取免费的试用账号吧!
使用方法
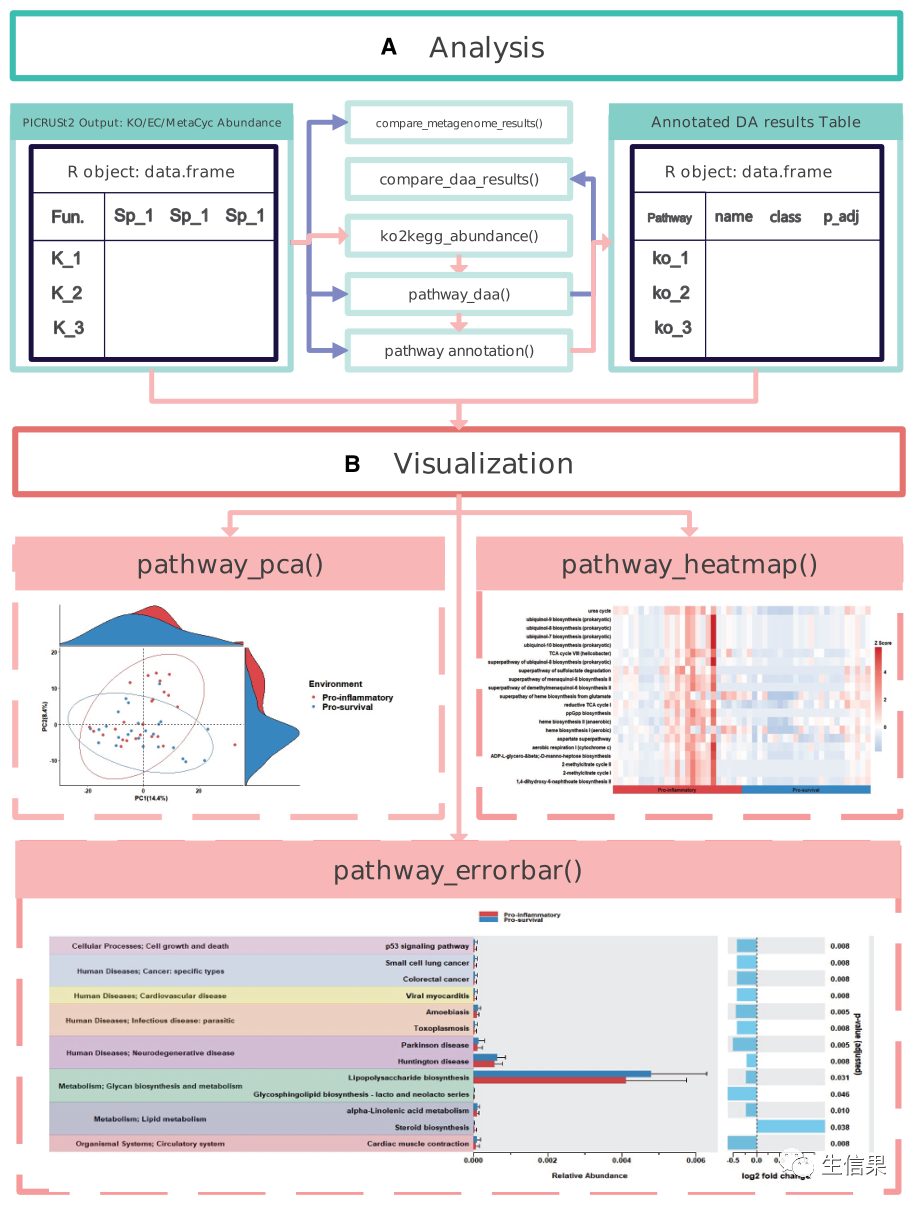
图3 分析流程(图源:Chen et al. 2023)
环境准备
安装和其他包一样,install.packages或者install_github。
install.packages("ggpicrust2")# Install the devtools package if not already installed# Install ggpicrust2 from GitHubdevtools::install_github("cafferychen777/ggpicrust2")
除了安装它自己以外还需要安装相关的依赖包。
if (!requireNamespace("BiocManager", quietly =TRUE))install.packages("BiocManager")pkgs <-c("phyloseq", "ALDEx2", "SummarizedExperiment", "Biobase", "devtools","ComplexHeatmap", "BiocGenerics", "BiocManager", "metagenomeSeq","Maaslin2", "edgeR", "lefser", "limma", "KEGGREST", "DESeq2")for (pkg in pkgs) {if (!requireNamespace(pkg, quietly =TRUE))BiocManager::install(pkg)}
 数据输入
数据输入
ggpicrust2建议采用PICRUSt2原始输出pred_metagenome_unstrat.tsv的数据格式,不过csv和txt也可以。
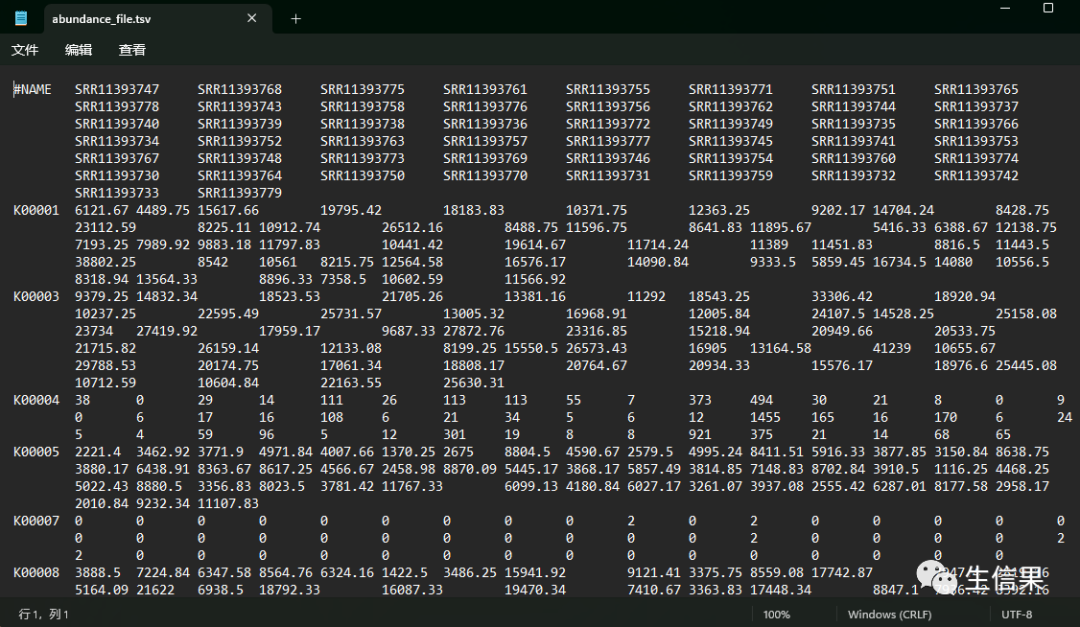
图4 输入数据
一个合格的包就得提供样例输入,这个包当然也不例外(小果分享的包都是优秀的好包!),作者不仅在包里提供了样例,还有单独的样例数据文件。链接如下:https://drive.google.com/drive/folders/1on4RKgm9NkaBCykMCCRvVJuEJeNVVqAF

图5 样例数据
有两种数据导入的方法,第一种是用包里的函数,第二种是用单独的读取文件函数,两种都很方便,看大家个人喜好,我认为第二种使用范围更广一些。
下面小果把代码奉上:
library(readr)library(ggpicrust2)library(tibble)# install.packages('tidyverse')library(tidyverse)library(ggprism)library(patchwork)path <-'D:/实习/ggpicrust2'setwd(path)# 数据文件下载地址# https://drive.google.com/drive/folders/1on4RKgm9NkaBCykMCCRvVJuEJeNVVqAF# 加载两个必须的数据: abundance data and metadata# 从文件读取abundance_file <-"abundance_file.tsv"metadata <-read_delim("metadata.txt",delim ="t",escape_double =FALSE,trim_ws = TRUE)# 从文件名开始results_file_input <-ggpicrust2(file = abundance_file,metadata = metadata,group ="Environment", # For example dataset, group = "Environment"pathway ="KO",daa_method ="LinDA",ko_to_kegg =TRUE,order ="pathway_class",p_values_bar =TRUE,x_lab ="pathway_name")# 看结果results_file_input[[1]]$plotresults_file_input[[1]][[1]]$results# 用导进来的数据,dataframe格式abundance_data <-read_delim(abundance_file, delim ="t", col_names =TRUE, trim_ws =TRUE)# 从读进来的数据开始results_data_input <-ggpicrust2(data = abundance_data,metadata = metadata,group ="Environment", # For example dataset, group = "Environment"pathway ="KO",daa_method ="LinDA",ko_to_kegg =TRUE,order ="pathway_class",p_values_bar =TRUE,x_lab ="pathway_name")results_data_input[[1]]$plotresults_data_input[[1]]$results
这里用到了这个包最主要的函数,也就是ggpicrust2(),这个函数继承了通路注释、10种最先进的差异丰度(DA)方法,以及DA的可视化结果。下面是小果为大家整理的参数介绍:
•file:字符串,表示包含KO丰度数据的输入文件的文件路径,其格式符合picrust2导出格式。输入文件应该在第一列包含KO标识符,在第一行包含样本标识符。剩余的单元格应该包含每个KO-样本对的丰度值。
•data:data.frame,以与输入文件相同的格式包含KO丰度数据。如果提供了此参数,函数将使用此数据而不是从文件中读取。默认情况下,此参数设置为NULL。
•metadata:tibble,包含样本信息。
•group:字符,组的名称。
•pathway:字符,”EC”、”KO”或”MetaCyc”之一。
•daa_method:字符,指定差异丰度分析的方法,选项有:
–”ALDEx2″:用于高通量测序数据的类似ANOVA的差异表达工具。
–”DESeq2″:基于负二项分布的差异表达分析,使用DESeq2。
–”edgeR”:使用edgeR对两组负二项分布计数之间的差异进行精确检验。
–”limma voom”:Limma-voom框架用于RNA-seq数据分析。
–”metagenomeSeq”:使用metagenomeSeq拟合逻辑回归模型,检验组之间的差异丰度。
–”LinDA”:用于微生物组成数据的差异丰度分析的线性模型。
–”Maaslin2″:用于多变量线性模型的多变量相关性分析,用于差异丰度分析。
•ko_to_kegg:字符,控制KO丰度到KEGG丰度的转换。
•p.adjust:字符,指定p值调整方法,选项有:
–”BH”:Benjamini-Hochberg校正。
–”holm”:Holm校正。
–”bonferroni”:Bonferroni校正。
–”hochberg”:Hochberg校正。
–”fdr”:False discovery rate校正。
–”none”:不进行p值校正。
•order:字符,控制主绘图行的顺序。
•p_values_bar:字符,控制主绘图是否具有p值条。
•x_lab:字符,控制x轴标签名称,可从”feature”、”pathway_name”和”description”中选择。
•select:包含要选择的通路名称的向量。
•reference:字符,用于几种DA方法的参考组水平。
•colors:一个包含颜色数量的向量。
下面就是DA的结果啦,包含了对应通路的相对丰度以及fold change。

图6 DA结果
因为ggpicrust2()是最主要的函数,它也会时不时有bug,所以作者告诉我们,如果有报错,可以试试下面这个流程。小果先把代码贴在下面:
# 加载MetaCyc pathway abundance 和 metadatadata("metacyc_abundance") # 加载MetaCyc通路丰度数据data("metadata") # 加载元数据# 如果你没有使用示例数据集,请将column_to_rownames()更改为适当的特征列# 如果你没有使用示例数据集,请将group更改为"你的分组列名"# 使用LinDA方法metacyc_daa_results_df <-pathway_daa(abundance = metacyc_abundance %>%column_to_rownames("pathway"), metadata = metadata, group ="Environment", daa_method ="LinDA")# 对不进行KO到KEGG转换的MetaCyc通路结果进行注释metacyc_daa_annotated_results_df <-pathway_annotation(pathway ="MetaCyc", daa_results_df = metacyc_daa_results_df, ko_to_kegg =FALSE)# 生成通路误差柱状图pathway_errorbar(abundance = metacyc_abundance %>%column_to_rownames("pathway"), daa_results_df = metacyc_daa_annotated_results_df, Group = metadata$Environment, ko_to_kegg =FALSE, p_values_threshold =0.05, order ="group", select =NULL, p_value_bar =TRUE, colors =NULL, x_lab ="描述")# 生成通路热图# install.packages('ggh4x')library(ggh4x)feature_with_p_0.05<- metacyc_daa_results_df %>%filter(p_adjust <0.05)pathway_heatmap(abundance = metacyc_abundance %>%filter(pathway %in% feature_with_p_0.05$feature) %>%column_to_rownames("pathway"), metadata = metadata, group ="Environment")# 生成通路PCA图pathway_pca(abundance = metacyc_abundance %>%column_to_rownames("pathway"), metadata = metadata, group ="Environment")# 对多个方法运行pathway_daamethods <-c("ALDEx2", "DESeq2", "edgeR")daa_results_list <-lapply(methods, function(method) {pathway_daa(abundance = metacyc_abundance %>%column_to_rownames("pathway"), metadata = metadata, group ="Environment", daa_method = method)})# 比较不同方法的结果comparison_results <-compare_daa_results(daa_results_list = daa_results_list, method_names =c("ALDEx2_Welch's t检验", "ALDEx2_Wilcoxon秩和检验", "DESeq2", "edgeR"))
这些函数的参数和ggpicrust2()类似,大家只要对照小果前文的参数解释即可。
PICRUSt2的主流可视化是bar_plot、error_bar_plot、pca_plot、heatmap_plot。pathway_errorbar()可以显示组间的相对丰度差异,以及DA结果的log2 fold change和p值。pathway_pca()可以通过主成分分析显示降维后的差异。pathway_heatmap()可以可视化PICRUSt2输出数据中的模式,这对于我们识别趋势或突出显示感兴趣的区域非常有用。
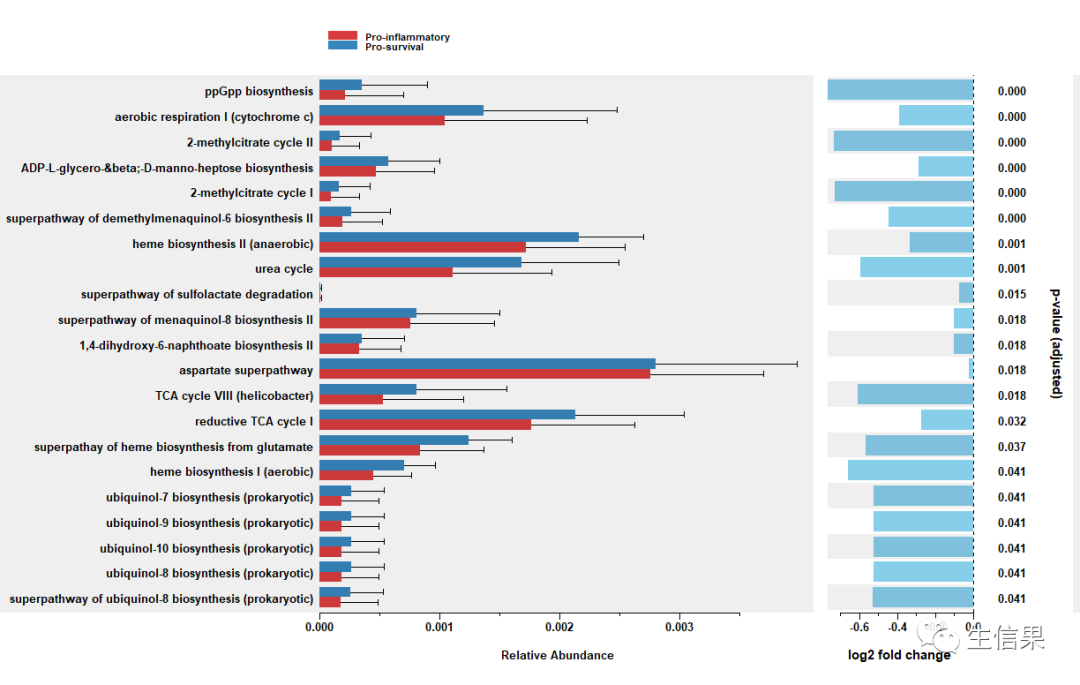
图7 bar plot

图8 热图
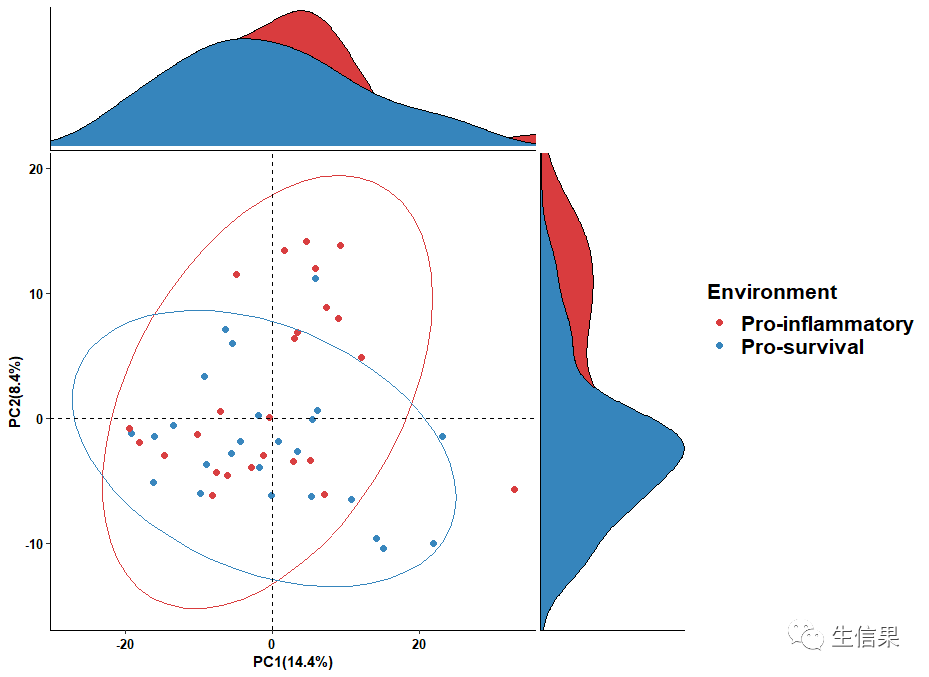
图9 PCA图
结语
总结一下。ggpicrust2是一个针对PICRUSt2功能预测的输出文件进行下游分析的R包,用于进行高级数据分析和结果的可视化。ggpicrust2已经被加入到PICRUSt2 wiki文档中,这反映了它在学术圈中得到了越来越多的认可和采用。
以上就是小果的分享啦!除了宏基因组功能预测,大家如果想筛选微生物的biomarker或者进行微生物群落划分,可以用我们云生信平台的小工具哦!
微生物群落划分、筛选微生物的biomarker
欢迎大家和小果一起讨论学习哦!我们下期再见~


往期推荐