探索细胞的奇妙之旅:splatter带你轻松玩转单细胞测序数据!
hello,你知道有一个R包可以生成模拟单细胞RNA-seq的数据,并可以用于评估和比较不同的分析方法嘛?今天小果就来教大家如何使用这个R包,感兴趣的话就和小果一起看下去吧!
了解splatter
splatter是一个用于模拟和分析单细胞转录组数据的R语言包。它提供了一系列功能,使研究人员可以生成模拟数据、进行参数估计、数据标准化、降维分析等。下面小果来介绍一下splatter包的主要功能和作用:
-
模拟数据生成:splatter允许用户根据自定义的参数生成模拟的单细胞测序数据。
-
参数估计:splatter提供了功能强大的参数估计方法,可以从给定的单细胞测序数据中估计出模型的参数。
-
数据标准化:splatter包含了一系列数据标准化方法,如对数转换、归一化等,可以帮助用户对单细胞测序数据进行预处理。
-
降维分析:splatter支持常用的降维分析方法,如主成分分析(PCA),可以将高维的单细胞测序数据投影到二维或三维空间中,方便可视化和进一步的数据解释。
-
数据可视化:splatter提供了多种绘图函数,用于可视化模拟数据、参数估计结果、标准化后的数据、降维分析结果等。
总而言之,splatter是一个功能丰富的R包,通过模拟生成、参数估计、数据标准化和降维分析等功能,帮助研究人员更好地理解和分析单细胞转录组数据,为单细胞研究提供了有力的工具和方法。
接下来小果将使用R语言中的splatter和scater包进行单细胞测序数据模拟和分析。那就和小果一起学起来吧!

1. 快速创建模拟数据
首先我们使用splatter包中的mockSCE函数快速创建一个模拟的单细胞测序数据。mockSCE函数可以设置细胞的数量(ncells)、基因的数量(ngenes)和spike-in的数量(nspikes),并返回一个SingleCellExperiment(SCE)对象。代码中的set.seed(1)用于设置随机数种子,保证结果的可重复性。最后,我们使用counts函数查看模拟数据的基因表达矩阵。
library(splatter)library(scater)### 1. 快速创建模拟数据# Usage# mockSCE(ncells = 200, ngenes = 2000, nspikes = 100)set.seed(1)sce <- mockSCE() # class: SingleCellExperimentcounts(sce)
我们一起来看一下模拟生成的基因表达矩阵:

2. 参数估计
接下来,我们使用splatEstimate函数从给定的数据中(这里是上一部分模拟生成的数据)估计参数。
### 2. 参数估计# 从数据(模拟数据或真实的单细胞测序数据)中估计参数params <- splatEstimate(sce) # Params object
3. 自定义参数,创建模拟数据
接下来,我们来学习如何根据自定义参数创建模拟数据。首先,使用newSplatParams函数创建一个参数对象params。然后,使用getParam函数查看参数对象中的参数设定。接下来,使用setParam和setParams函数设置参数对象中的参数。最后,使用splatSimulate函数根据参数对象创建模拟数据。
### 3. 自定义参数,创建模拟数据# 创建参数对象params <- newSplatParams() #默认10000 Genes,100 Cells# 查看参数对象中的参数设定getParam(params, "nGenes")getParam(params, "nCells")getParams(params, c("nGenes", "mean.rate", "mean.shape"))# 设定参数对象中的参数params <- setParam(params, "nGenes", 5000)params <- setParam(params, "batchCells", 50)params <- setParams(params, mean.shape = 0.5, de.prob = 0.2)params <- setParams(params, update = list(nGenes = 8000, mean.rate = 0.5))
# 根据参数,创建模拟数据sim <- splatSimulate(params)

4. 提取SingleCellExperiment类信息
接下来,我们就可以介绍提取SingleCellExperiment对象中的信息。
### 4. 提取SingleCellExperiment类信息class(sim)# Information about geneshead(rowData(sim))# Information about cellshead(colData(sim))# Gene by cell matricesnames(assays(sim))# Example of cell means matrixassays(sim)$CellMeans[1:5, 1:5]# 表达矩阵counts <- counts(sim)counts[1:3,1:5]class(counts) # "matrix" "array"typeof(counts) # [1] "integer"dim(counts) #[1] 8000 100
运行结果如下:

5. 数据标准化,对数化和降维
进一步地,我们就可以如单细胞测序数据进行标准化、对数化和降维处理。首先,使用logNormCounts函数对SingleCellExperiment对象中的counts值进行对数转换和归一化。通过logcounts函数可以查看转换后的对数表达矩阵。然后,使用runPCA函数对数据进行PCA降维。
### 5. 数据标准化,对数化和降维## {scuttle}包的函数:logNormCounts## {scater}包的函数:runPCA,plotPCA# SingleCellExperiment 对象的counts值计算对数转换的归一化表达式值sim <- logNormCounts(sim)counts(sim)[1:3,1:5]logcounts(sim)[1:3,1:5]# PCA降维sim <- runPCA(sim)plotPCA(sim)
最后,小果使用plotPCA函数绘制PCA降维结果。我们来看看可视化的结果是什么样吧!
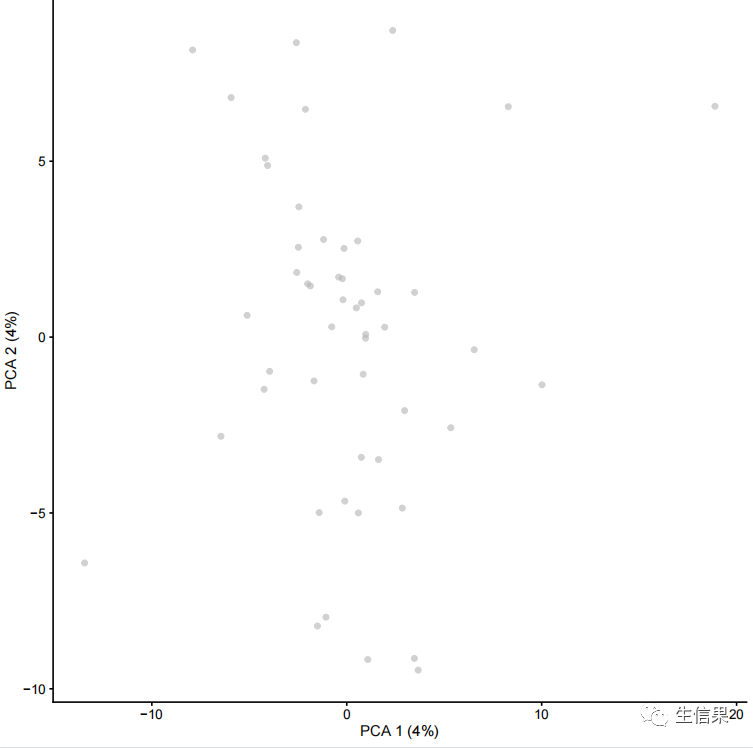
6. 创建复杂的模拟数据
最后,我们可以创建更复杂的模拟数据,包括具有组结构、路径结构和批次效应的模拟数据。
### 6. 创建复杂的模拟数据## groupssim.groups <- splatSimulate(group.prob = c(0.3, 0.7), method = "groups",verbose = FALSE)sim.groups <- logNormCounts(sim.groups)sim.groups <- runPCA(sim.groups)plotPCA(sim.groups, colour_by = "Group")
我们来看看降维后的结果:

dim(counts(sim.groups))rowData(sim.groups)$DEFacGroup1rowData(sim.groups)$DEFacGroup2metadata(sim.groups)## pathssim.paths <- splatSimulate(de.prob = 0.2, nGenes = 1000, method = "paths",verbose = FALSE)sim.paths <- logNormCounts(sim.paths)sim.paths <- runPCA(sim.paths)plotPCA(sim.paths, colour_by = "Step")colData(sim.paths)$Step## batchessim.batches <- splatSimulate(batchCells = c(50, 50), verbose = FALSE)sim.batches <- logNormCounts(sim.batches)sim.batches <- runPCA(sim.batches)plotPCA(sim.batches, colour_by = "Batch")rowData(sim.batches)$BatchFacBatch1rowData(sim.batches)$BatchFacBatch2dev.off()
最后pca可视化的结果如下:
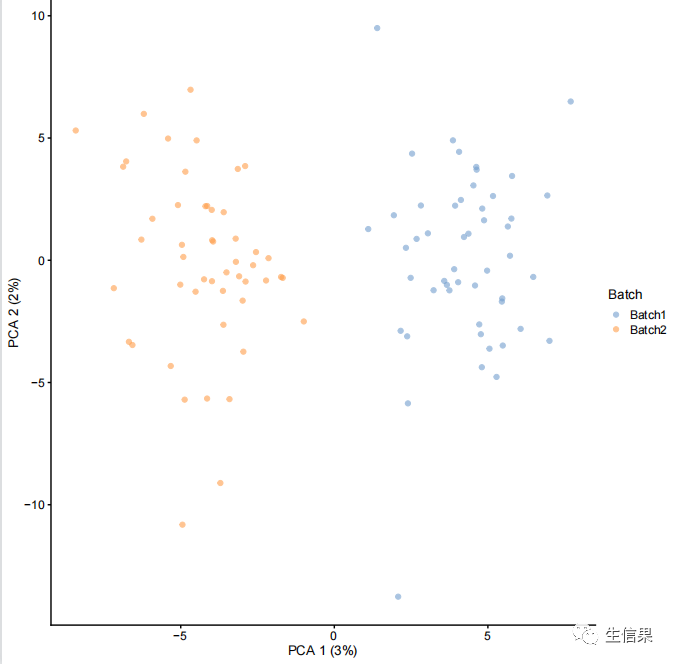
通过学习这些代码,你可以了解如何使用splatter和scater包进行单细胞测序数据的模拟和分析,包括模拟数据的快速生成、参数估计、自定义参数创建、数据提取、数据标准化和降维处理等。怎么样,是不是很简单,你学废了嘛!?
往期推荐

