使用R语言包genefilter预处理数据和筛选数据的保姆级教程
在生物学研究中,我们经常需要处理大规模的基因表达数据,这些数据可能来自不同条件下的实验或疾病样本。genefilter提供了一套功能强大的工具,用于处理这些复杂的数据,并从中识别出与研究问题相关的基因。
要使用genefilter包,可以在R中使用以下命令进行安装和加载:
> install.packages("genefilter")> library(genefilter)
genefilter的一项主要功能是基因表达数据的预处理。预处理是数据分析的关键步骤之一,它可以消除数据中的噪声、处理缺失值,并对数据进行归一化,以确保数据的准确性和可靠性。这对于基因表达数据尤为重要,因为在实验中,数据可能会受到多种因素的影响,例如技术平台的差异、实验条件的变化等。genefilter提供了多种方法来完成这些任务,帮助研究人员获得高质量的基因表达数据。
示例:
当使用R中的genefilter包进行基因表达数据的预处理时,我们通常会涉及数据的读取、数据清洗、归一化和可视化。下面是一个简单的基因表达数据预处理示例,使用R中自带的iris数据集。

# 安装和加载所需的包> install.packages("genefilter")> install.packages("Biobase") # 如果未安装Biobase包的话> library(genefilter)> library(Biobase)# 使用iris数据集作为示例数据> data(iris)# 将iris数据集的前四列作为基因表达数据(示例中并不是真实基因表达数据)> gene_expr <- iris[, 1:4]# 查看原始数据的前几行> head(gene_expr)Sepal.Length Sepal.Width Petal.Length Petal.Width1 5.1 3.5 1.4 0.22 4.9 3.0 1.4 0.23 4.7 3.2 1.3 0.24 4.6 3.1 1.5 0.25 5.0 3.6 1.4 0.26 5.4 3.9 1.7 0.4# 归一化# 在基因表达数据中,归一化是为了使不同基因的表达值在数量级上保持一致,便于比较。> normalized_data <- scale(gene_expr)# 查看归一化后的数据前几行> head(normalized_data)Sepal.Length Sepal.Width Petal.Length Petal.Width[1,] -0.8976739 1.01560199 -1.335752 -1.311052[2,] -1.1392005 -0.13153881 -1.335752 -1.311052[3,] -1.3807271 0.32731751 -1.392399 -1.311052[4,] -1.5014904 0.09788935 -1.279104 -1.311052[5,] -1.0184372 1.24503015 -1.335752 -1.311052[6,] -0.5353840 1.93331463 -1.165809 -1.048667# Boxplot> boxplot(normalized_data, col = "lightblue", main = "Normalized Gene Expression",+ xlab = "Gene", ylab = "Normalized Expression")
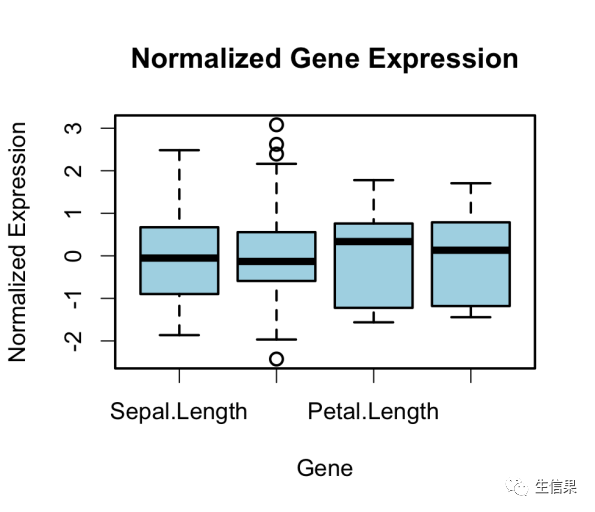
> install.packages("genefilter")> library(genefilter)

# 利用可视化图表,我们可以初步观察基因表达数据的分布情况和基因之间的相关性。# 然后,我们可以根据实际研究问题和需要,进一步使用genefilter包提供的功能,筛选出感兴趣的基因并进行深入分析。除了数据预处理外,genefilter还提供了丰富的筛选方法,用于从大规模基因表达数据中挑选出与研究问题最相关的基因。这些筛选方法可以根据基因的表达模式、变异程度、相关性等指标进行筛选,并且可以根据用户的需求进行定制。通过这些筛选方法,研究人员可以快速地从海量的基因表达数据中找到那些对于研究问题最有意义的基因,从而缩小研究范围,提高研究效率。示例:airway数据集包含了来自人类气道上皮细胞的RNA-seq数据,用于研究哮喘病的基因表达变化。我们将使用genefilter包中的筛选方法来根据基因的表达模式和变异程度进行筛选,并通过可视化图表来展示结果。# 安装和加载所需的包> install.packages("genefilter")> install.packages("airway") # 如果未安装airway包的话> library(genefilter)> library(airway)# 加载airway数据集> data(airway)# 提取基因表达数据,假设数据是矩阵形式,行为基因,列为样本> gene_expr <- assay(airway)# 查看原始数据的前几行> head(gene_expr)SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521ENSG00000000003 679 448 873 408 1138 1047 770 572ENSG00000000005 0 0 0 0 0 0 0 0ENSG00000000419 467 515 621 365 587 799 417 508ENSG00000000457 260 211 263 164 245 331 233 229ENSG00000000460 60 55 40 35 78 63 76 60ENSG00000000938 0 0 2 0 1 0 0 0# 筛选方法一:根据基因的表达模式进行筛选# 使用genefilter包中的`rowSds`函数计算每个基因的标准差(表达模式),# 然后选择标准差大于指定阈值的基因作为筛选结果。> threshold_sd <- 1 # 设置标准差的阈值> selected_genes_by_sd <- gene_expr[rowSds(gene_expr) > threshold_sd, ]# 可视化:箱线图比较筛选前后的数据> par(mfrow = c(1, 2))> boxplot(gene_expr, col = "lightblue", main = "Before Filtering", xlab = "Gene", ylab = "Expression")> boxplot(selected_genes_by_sd, col = "lightgreen", main = "After Filtering by SD", xlab = "Gene", ylab = "Expression")
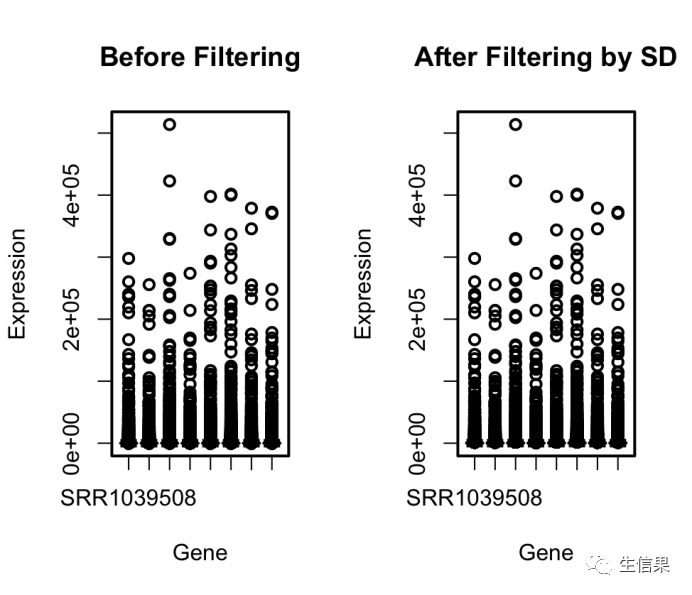
# 筛选方法二:根据基因的变异程度进行筛选# 使用genefilter包中的`rowVars`函数计算每个基因的方差(变异程度),# 然后选择方差大于指定阈值的基因作为筛选结果。> threshold_var <- 0.1 # 设置方差的阈值> selected_genes_by_var <- gene_expr[rowVars(gene_expr) > threshold_var, ]# 可视化:箱线图比较筛选前后的数据> par(mfrow = c(1, 2))> boxplot(gene_expr, col = "lightblue", main = "Before Filtering", xlab = "Gene", ylab = "Expression")> boxplot(selected_genes_by_var, col = "lightgreen", main = "After Filtering by Variance", xlab = "Gene", ylab = "Expression")

在这个示例中,我们使用了两种筛选方法:基于标准差(表达模式)和基于方差(变异程度)。通过可视化箱线图,我们可以直观地比较筛选前后的基因表达数据分布。通过调整阈值,我们可以灵活地选择适合特定研究问题的基因。
genefilter还支持并行计算,这使得处理大规模数据变得更加高效。在处理大规模基因表达数据时,计算复杂度可能非常高,使用并行计算可以充分利用计算机资源,加速数据处理过程。
以上就是对R语言包genefilter的简单介绍啦,它提供了数据预处理和筛选功能,帮助研究人员从海量的基因表达数据中挖掘出有意义的信息,为生物学研究提供有力的支持。无论是在生物医学研究、生物信息学分析还是基因组学研究领域,genefilter都是一个非常有价值的工具,有助于加快研究进展,促进生物学领域的发展。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~
或者也可以关注我们的官网也会持续更新的哦~
往期推荐

