bed文件处理工具之——bedtools

大家好!小果我呀,今天我要给大家推荐一个超好用的生信小工具——bedtools!
bedtools是一个处理基因组bed文件的软件包,它可以实现各种基因组分析,比如寻找重叠区域、计算覆盖度、合并或切分区间等等。bedtools的特点是它支持多种文件格式,比如BED、GFF、VCF等,而且它的操作也非常简单,只需要几个命令就可以完成复杂的任务。bedtools还可以和其他工具配合使用,比如samtools、bowtie2、bwa等,从而实现更多的功能。bedtools是一个非常实用的工具,强烈推荐大家学习和使用它。
那么,怎么安装和使用bedtools呢?其实很简单,只需要三步就可以了。
第一步,下载bedtools的源码包,可以从官网或者GitHub(https://github.com/arq5x/bedtools2 )上获取。
第二步,解压源码包,并在终端中进入解压后的文件夹。
unzip bedtools2-master.zipcd bedtools2-master/
第三步,输入make命令,编译和安装bedtools。这样就完成了bedtools的安装啦!非常方便吧?
接下来,给大家演示一下bedtools的基本用法。

首先,我们需要准备一些输入文件,比如两个BED文件(此例子中的bed文件来自bedtools自带的/test/ 文件夹),分别表示两组基因组区域。我们可以用cat命令查看一下这两个文件的内容:
$ cat A.bedchr1 10 20 a1 1 +chr1 100 200 a2 2 -$ cat B.bedchr1 20 30 b1 1 +chr1 90 101 b2 2 -chr1 100 110 b3 3 +chr1 200 210 b4 4 +samtools view test.bam@HD VN:1.0 GO:none SO:coordinate@SQ SN:chr1 LN:249250621paired-1 1 chr1 1 40 30M * 0 0 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA DDDDDDDDDDDDDDDDDDDDDDDDDDDDDD MD:Z:50paired-1 1 chr1 1 100 30M * 0 0 GGGGGGGGGGGGGGGGGGGGGGGGGGGGGG EEEEEEEEEEEEEEEEEEEEEEEEEEEEEE MD:Z:50unpaired 1 chr1 1 100 30M * 0 0 GAAGGCCACCGCCGCGGTTATTTTCCTTCA CCCDDB?=FJIIJIGFJIJHIJJJJJJJJI MD:Z:50paired-2 1 chr1 1 40 30M * 0 0 TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT FFFFFFFFFFFFFFFFFFFFFFFFFFFFFF MD:Z:50
1. Bedtools之intersect:寻找这两个文件的重叠区域。
Usage: bedtools intersect [OPTIONS] -a <bed/gff/vcf/bam> -b <bed/gff/vcf/bam>$ bedtools intersect -a a.bed -b b.bedchr1 100 101 a2 2 -chr1 100 110 a2 2 -这样就得到了重叠区域的坐标,非常快速和准确。我们还可以用-wa和-wb选项来输出完整的重叠区域信息:$ bedtools intersect -a A.bed -b B.bed -wa -wbABchr1 100 200 a2 2 - chr1 90 101 b2 2 -chr1 100 200 a2 2 - chr1 100 110 b3 3 +这样就可以看到每个重叠区域分别属于哪两个输入文件。我们还可以用-loj选项来输出左连接的结果,即输出所有A文件中的区域,以及与之重叠或者不重叠的B文件中的区域:$ bedtools intersect -a A.bed -b B.bed -lojchr1 10 20 a1 1 + . -1 -1 . -1 .chr1 100 200 a2 2 - chr1 90 101 b2 2 -chr1 100 200 a2 2 - chr1 100 110 b3 3 +
2. Bedtools之merge:可以将一个BED文件中所有的重叠区域和相邻merge为一个大的区域。命令如下:
$bedtools merge -i B.bedchr1 20 30chr1 90 110chr1 200 210输出的三列为:染色体、合并区间的起始位置和结束位置。B注意:使用 merge 命令之前,需要确保输入文件已经按照染色体顺序排序,这样可以加快处理速度并减少内存占用。如果输入文件没有排序,bedtools merge 可能会运行出错。
3. bedtools之格式转换
bedtools可以做以下几种格式转换:bamtobed、bedtobam、bamtofastq、bedpetobam、bed12tobed6,这里就以bamtobed为例给大家演示以下吧。$ bedtools bamtobed -i test.bamchr1 0 30 paired-1 40 +chr1 0 30 paired-1 100 +chr1 0 30 unpaired 100 +chr1 0 30 paired-2 40 +
4. Bedtools之coverage:计算深度和广度。
Bedtools coverage可以计算BED文件B在A文件上的覆盖深度和广度。命令如下:
$ bedtools coverage -a A.bed -b B.bed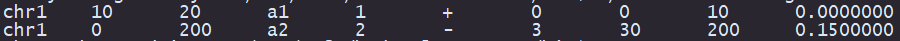
5. bedtools之map
bedtools map 的功能是将一个文件中的区域(例如基因或转录本)映射到另一个文件中的区域(例如测序读取或峰值),并计算每个区域的覆盖度或其他统计量。这个工具可以用来分析不同样本之间的区域重叠情况,或者评估某些特征(例如甲基化或组蛋白修饰)在特定区域的分布情况。bedtools map 的使用方法如下:
bedtools map -a A.bed -b B.bed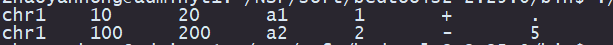
当然,bedtools还有很多其他的命令和选项,比如complement、subtract、sort、shuffle等等,我就不一一介绍了,大家可以自己去探索和尝试哦!


