超详细的差异表达分析热图可视化代码解析
点击蓝字 关注我们
差异表达分析的热图是通过可视化差异表达基因的表达模式来揭示基因调控网络中的重要变化。绘制差异基因的热图。热图可以帮助发现在不同条件下显著差异表达的基因。通过设置差异表达分析的阈值,例如logFC(fold change)和p值或FDR(false discovery rate),筛选出显著差异表达的基因,并在热图中突出显示这些基因。这有助于快速鉴定与特定生物学条件或疾病状态相关的差异表达基因。
下面小花带你用vcan基因进行操作,数据使用tcga下载的你要研究的癌种数据矩阵就可以。
#if (!requireNamespace("BiocManager", quietly = TRUE))# install.packages("BiocManager")#BiocManager::install("limma")#install.packages("pheatmap")安装limma和pheatmap。首先检查是否已经安装了BiocManager包,#引用包library(limma)library(pheatmap)加载所需的R包,以便在代码中使用相应的函数和功能。gene="VCAN" #示例目标基因的名称expFile="symbol.txt" #表达输入文件logFCfilter=1 #logFC过滤条件fdrFilter=0.05 #fdr过滤条件setwd("C:\biowolf\Gene\19.diff") #设置工作目录指定目标基因的名称、表达输入文件的名称、logFC过滤条件和fdr过滤条件。另外,通过setwd函数设置R的工作目录#读取文件,并对输入文件整理rt=read.table(expFile, header=T, sep="t", check.names=F)原始基因表达量数据symbol.xt
rt=as.matrix(rt)rownames(rt)=rt[,1]exp=rt[,2:ncol(rt)]dimnames=list(rownames(exp), colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp), dimnames=dimnames)data=avereps(data)
#输入文件中读取表达数据,并对数据进行整理。表达数据是基因的表达量数据,#读取后转换为矩阵形式,并设置行名和列名。使用avereps函数对数据进行平#均处理,处理后的数据存储在data变量中。#去除正常样品group=sapply(strsplit(colnames(data),"\-"), "[", 4)group=sapply(strsplit(group,""), "[", 1)group=gsub("2", "1", group)data=data[,group==0]data=t(data)rownames(data)=gsub("(.*?)\-(.*?)\-(.*?)\-(.*?)\-.*","\1\-\2\-\3", rownames(data))rt=t(avereps(data))#根据样品名称中的编号信息,将正常样品和其他样品分开。通过正则表达#式操作,将样品名中的部分信息提取出来,并去除样品编号中的"2"。然后,#根据样品名中的信息重新命名行名,并将数据重新转置。#按照基因表达的中位值对样品分组low=rt[gene,]<=median(rt[gene,])high=rt[gene,]>median(rt[gene,])lowRT=rt[,low]highRT=rt[,high]conNum=ncol(lowRT) #低表达组样品数目treatNum=ncol(highRT) #高表达组样品数目data=cbind(lowRT,highRT)Type=c(rep(1,conNum), rep(2,treatNum))#根据目标基因的表达值中位数,将样品分为低表达组和高表达组。#根据低表达组和高表达组的列数,分别存储在lowRT和highRT变量中。3conNum和treatNum分别表示低表达组和高表达组的样品数目。#两个组的数据合并为一个新的数据矩阵,用Type向量表示每个样品所属组。#差异分析outTab=data.frame()for(i in row.names(data)){rt=data.frame(expression=data[i,], Type=Type)wilcoxTest=wilcox.test(expression ~ Type, data=rt)conGeneMeans=mean(data[i,1:conNum])treatGeneMeans=mean(data[i,(conNum+1):ncol(data)])logFC=log2(treatGeneMeans)-log2(conGeneMeans)pvalue=wilcoxTest$p.valueconMed=median(data[i,1:conNum])treatMed=median(data[i,(conNum+1):ncol(data)])diffMed=treatMed-conMedif( ((logFC>0) & (diffMed>0)) | ((logFC<0) & (diffMed<0)) ){outTab=rbind(outTab,cbind(gene=i,lowMean=conGeneMeans,highMean=treatGeneMeans,logFC=logFC,pValue=pvalue))}}pValue=outTab[,"pValue"]fdr=p.adjust(as.numeric(as.vector(pValue)), method="fdr")outTab=cbind(outTab, fdr=fdr)#对于每个基因,通过Wilcoxon秩和检验比较低表达组和高表达组之间的差异。#计算低表达组和高表达组的基因均值、logFC、p值和差异中值。#根据logFC的方向和差异中值的方向,将符合条件的基因结果添加到outTab数据框中。#pValue是p值的向量,使用p.adjust函数计算多重检验校正后的fdr值,将fdr#值添加到outTab中。#输出差异的结果outDiff=outTab[( abs(as.numeric(as.vector(outTab$logFC)))>logFCfilter & as.numeric(as.vector(outTab$fdr))write.table(outDiff, file="diff.txt", sep="t", row.names=F, quote=F)#输出符合过滤条件的差异基因结果。根据设定的logFC过滤条件和fdr过滤条件,#筛选出差异基因,并将结果写入到名为"diff.txt"的文件中。
#绘制差异基因的热图geneNum=50 #设置显示基因的数目outDiff=outDiff[order(as.numeric(as.vector(outDiff$logFC))),]diffGeneName=as.vector(outDiff[,1])diffLength=length(diffGeneName)hmGene=c()if(diffLength>(2*geneNum)){hmGene=diffGeneName[c(1:geneNum,(diffLength-geneNum+1):diffLength)]}else{hmGene=diffGeneName}hmExp=log2(data[hmGene,]+0.01)Type=c(rep("Low",conNum),rep("High",treatNum))Type=factor(Type, levels=c("Low","High"))names(Type)=colnames(data)Type=as.data.frame(Type)pdf(file="heatmap.pdf", width=10, height=7)#绘制差异基因的热图。首先,根据设定的基因数目,选择要显示的差异基因名#称。根据选择的差异基因,从原始数据中提取相应的表达值,并进行log2转换。#Type向量表示每个样品所属的组,根据样品的分组情况创建一个数据框。接下#来,使用pheatmap函数绘制热图。设置热图的参数,包括注释、颜色渐变、#是否进行列聚类、是否显示列名、行缩放等。将绘制的热图保存为PDF文件。pheatmap(hmExp,annotation=Type,color = colorRampPalette(c(rep("blue",5), "white", rep("red",5)))(50),cluster_cols =F,show_colnames = F,scale="row",fontsize = 8,fontsize_row=5,fontsize_col=8)dev.off()
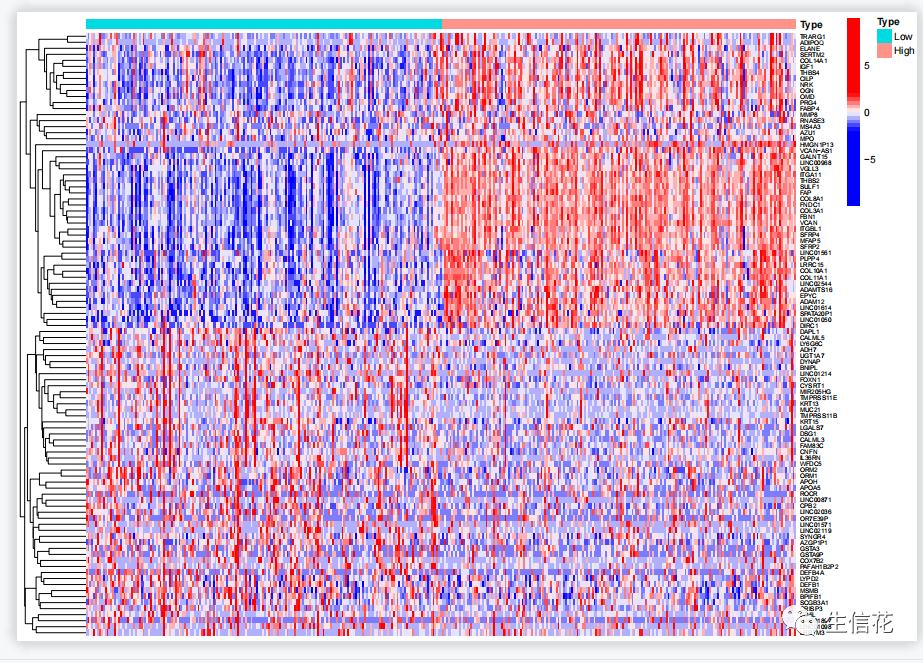
差异表达分析的热图具有可视化差异基因表达模式、发现差异基因、研究基因调控网络和生物过程、指导生物学研究和实验设计,以及可视化和报告结果的功能和意义。它在生物信息学和生物医学研究中起着重要的作用。下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
欢迎使用:云生信 – 学生物信息学 (biocloudservice.com)
如果想用服务器可以联系微信:18502195490(快来联系我们使用吧!)


(点击阅读原文跳转)
![]() 点一下阅读原文了解更多资讯
点一下阅读原文了解更多资讯
