小果五分钟教会你如何使用R语言包scuttle进行RNA数据分析

R语言作为一种广泛用于数据分析和统计建模的编程语言,拥有丰富的扩展包,以满足各种学科领域的需求。在生物学领域,研究人员常常需要处理大规模的生物信息数据,例如基因表达数据、蛋白质互作网络、代谢通路等。为了更有效地进行生物信息学分析,R语言社区开发了众多生物信息学相关的包,其中之一便是scuttle。
scuttle是R语言中备受欢迎的生物信息学包之一,其特色功能在于支持单细胞RNA测序(scRNA-seq)数据的深入分析和直观可视化。单细胞RNA测序作为一项先进的技术,已经引发了生物学领域的巨大关注。它突破了传统基因表达测定的限制,能够在单个细胞的水平上测量基因的表达水平,从而为我们展示了细胞在转录组水平上的多样性和差异。然而,这种高分辨率的数据也带来了分析的挑战,需要强大的工具来揭示数据中蕴藏的宝贵信息。

正是在这种背景下,scuttle崭露头角。作为一个专注于单细胞RNA测序数据的R包,scuttle旨在协助生物学家和生物信息学家解析这些复杂的数据,为他们提供了一套强大的分析和可视化工具,帮助深入了解细胞的多样性、发育过程和疾病机制。通过scuttle,我们能够在细胞的微观层面上挖掘出前所未见的洞察力。
在这个多功能的包中,scuttle首先提供了数据的质量控制和预处理功能,帮助用户过滤掉低质量的细胞和基因,同时校正批次效应,确保后续分析的可靠性。这一步骤的关键性在于保障数据的准确性,为后续的探索打下坚实的基础。
I. 深入了解细胞多样性与发育过程
在现代生物学领域,单细胞RNA测序技术的兴起为我们打开了观察细胞内部工作机制的新视角。scuttle作为一款专注于单细胞RNA测序数据分析的R包,发挥着独特的优势。其显著功能之一便是通过聚类和降维分析帮助生物学家深入理解细胞的多样性和发育过程。在细胞的转录组水平上,不同类型的细胞可能表现出巨大的基因表达差异,这些差异往往直接关联着细胞的功能和特性。scuttle可以将高维的scRNA-seq数据转化为低维空间,如主成分分析(PCA)和t分布随机邻域嵌入(t-SNE)等方法,从而在二维或三维平面上呈现细胞的分布。这使得研究人员能够更准确地识别和理解细胞的聚类模式、不同细胞类型以及在发育过程中细胞状态的变化。
通过这种降维和可视化的手段,scuttle赋予了研究人员深入探索细胞多样性的能力。无论是在研究正常组织中的细胞异质性,还是在揭示组织发育过程中的细胞分化,scuttle都为生物学家提供了一个有力的工具。研究人员可以通过观察聚类图、t-SNE图以及其他降维可视化图表,更清晰地观察不同细胞的分布和趋势。这种深入的理解将为生物学家们构建细胞类型分类、揭示发育轨迹以及发现新的细胞亚群提供极大的帮助。
II. 揭示疾病机制和药物靶点
除了在探索正常生物学过程中的应用,scuttle在研究疾病机制和药物研发领域也扮演着重要角色。疾病往往表现为细胞状态的异常,而这些变化可能在基因表达水平上得到体现。通过比较病态细胞和正常细胞的基因表达水平,我们可以识别出与疾病发展密切相关的关键基因。scuttle的基因表达差异分析功能为研究人员提供了一个有效的方式来鉴定这些基因,从而深入了解疾病的分子机制。
在这个过程中,降维和可视化的技术也发挥着重要作用。通过绘制基因表达的热图、箱线图等图表,研究人员可以直观地展示不同基因在疾病和正常样本之间的差异。这不仅有助于验证差异表达基因的可靠性,还能够揭示它们在细胞类型和通路调控方面的潜在功能。这种分析和可视化的结合,为寻找潜在的治疗靶点和药物开发提供了有力的支持,为疾病研究带来了新的希望。
III. 揭示疾病的分子机制与发展
另一个使scuttle备受关注的领域是在疾病研究中的应用。疾病的发生往往伴随着细胞内基因表达的改变,而这些变化可以通过单细胞RNA测序技术来精确捕捉和分析。在这方面,scuttle展现出强大的基因表达差异分析功能,它为研究人员提供了一个有力工具,可以在细胞层面上识别不同状态下的基因表达差异。通过比较正常和异常细胞的基因表达,我们能够鉴定在特定疾病机制中起关键作用的基因,进而深入探究疾病的分子机制。
scuttle的基因表达差异分析不仅帮助我们发现在疾病中表达水平显著改变的基因,还能够帮助我们理解这些基因在疾病发展中的作用。通过对这些差异基因的功能、通路和相互关系的深入研究,我们可以揭示出疾病的潜在机制,为疾病的预防、诊断和治疗提供有力支持。特别是在寻找治疗靶点和开发新药方面,scuttle为研究人员提供了一个全面的视角,使他们能够更精准地干预疾病过程。
IV. 通过细胞层面的分析揭示疾病的复杂性
疾病往往是一个极其复杂的过程,涉及多种细胞类型、通路和信号调控网络的紊乱。通过scuttle的基因表达差异分析功能,我们能够将分子改变引起的细胞层面的响应更加详细地捕捉到。这为我们提供了一个深入研究疾病发展的窗口,让我们能够看到细胞内部的变化,理解这些变化如何影响整体的疾病过程。
更进一步地,scuttle的可视化功能使得这种细胞层面的分析更加直观和易于解释。通过绘制差异基因的热图、箱线图等图表,研究人员可以直观地展示这些基因在不同状态下的表达水平,揭示它们之间的关系和趋势。这种综合分析和可视化的方法,不仅有助于验证分析的可靠性,还能够为进一步的实验设计和机制探究提供有力的指导。
V. 强大的数据可视化能力
除了丰富的分析功能,scuttle的另一个引人注目的特点是其卓越的数据可视化能力。在生物信息学领域,数据的可视化是将分析结果传递给他人、发现模式并产生新的洞察力的关键途径。scuttle在这一领域发挥着重要作用,为用户提供了多种图表类型,如散点图、箱线图、热图等,使得复杂的数据和分析结果可以以直观的方式呈现,更容易被生物学家、研究人员和观众理解和解释。
通过可视化,用户可以直观地观察数据中的模式和趋势,从而更好地理解数据所蕴含的信息。例如,绘制散点图可以展示不同细胞的基因表达模式,揭示细胞之间的相似性和差异性。箱线图则能够清晰地显示基因表达的分布和差异,帮助用户识别显著的差异表达基因。另外,热图可以将基因的表达水平编码为颜色,将大量数据以直观的方式展示出来,帮助用户快速识别在不同细胞或条件下的表达模式。
VI. 促进信息提取和新假设的生成
数据可视化不仅仅是将数据展示出来,更是帮助用户从数据中提取有价值的信息、产生新的科学假设的过程。scuttle的图表不仅美观易懂,更能帮助用户将数据中的细微变化和关联关系快速捕捉到。例如,在基因表达差异分析中,通过绘制差异基因的箱线图,用户可以直观地发现在不同条件下基因表达的分布差异,进而推测这些基因在细胞过程中的潜在作用。
数据可视化还能够帮助研究人员从不同角度审视数据,产生新的研究假设。当我们将数据可视化为图表时,我们可能会注意到之前忽略的模式、趋势或异常。这种洞察力可以引导研究人员进一步深入探索,提出新的科学问题,以及设计更有针对性的实验方案。
要使用scuttle包,可以在R中使用以下命令进行安装和加载:
install.packages("scuttle") #安装scuttle语言包library(scuttle) #加载语言包scuttle的核心功能包括:
质量控制和预处理在单细胞RNA测序数据分析中扮演着至关重要的角色。由于技术噪声和批次效应的存在,这些数据往往需要经过严格的处理,以确保后续分析的准确性和可靠性。scuttle作为一款强大的单细胞数据分析工具,专注于解决这些挑战。
在质量控制方面,scuttle提供了一系列功能,可以帮助用户识别和排除低质量的细胞和基因。通过消除这些问题数据点,用户能够获得更干净、可靠的数据集,为后续分析奠定坚实的基础。此外,scuttle还能够校正批次效应,这是特别重要的,因为批次效应可能导致数据集中存在不必要的变异,影响对生物学现象的准确理解。
细胞聚类和降维分析是单细胞数据探索和理解的关键步骤。scuttle提供了一系列强大的聚类算法,使用户能够将相似的细胞归为同一类别,从而揭示出不同细胞类型之间的关系。此外,降维分析工具如主成分分析(PCA)和t分布随机邻域嵌入(t-SNE)能够将高维数据映射到二维空间,使用户能够在直观的图形中观察数据的分布和结构。
基因表达差异分析是了解不同细胞类型或条件下基因表达变化的关键。scuttle使用户能够比较不同情况下的基因表达水平,从而识别出在特定生物学过程中起关键作用的基因。这有助于深入理解细胞在不同条件下的功能和响应。
对于发育过程的研究,轨迹分析是一项强大的工具。scuttle能够帮助用户构建细胞状态的发展轨迹,从而揭示细胞的分化和演化过程。这对于理解生物学中动态的变化和发展至关重要。
scuttle内置了丰富的数据可视化功能,使用户能够以直观的方式展示他们的分析结果。无论是通过热图、散点图还是轨迹图,用户都能够更清晰地传达他们的发现,帮助其他研究人员更好地理解单细胞数据分析的结果。
示例:
当使用scuttle包进行单细胞RNA测序数据分析时,我们可以以一个简单的示例为例,使用一个包含细胞类型和基因表达信息的数据集。
安装和加载scuttle包install.packages("scuttle")library(scuttle)移除低基因表达的细胞scRNA_data_filtered <- filterCells(scRNA_data, min_genes = 200)校正批次效应scRNA_data_corrected <- correctBatchEffect(scRNA_data_filtered, batch_var = "Batch")进行PCA降维scRNA_data_pca <- runPCA(scRNA_data_corrected, ncomponents = 30)进行t-SNE降维scRNA_data_tsne <- runTSNE(scRNA_data_pca)根据细胞类型标识,进行基因表达差异分析de_genes <- differentialGeneTest(scRNA_data_corrected, grouping_var = "CellType")选取显著差异表达基因top_de_genes <- subset(de_genes, padj < 0.05)绘制细胞聚类的t-SNE图plotTSNE(scRNA_data_tsne, color_by = "CellType")
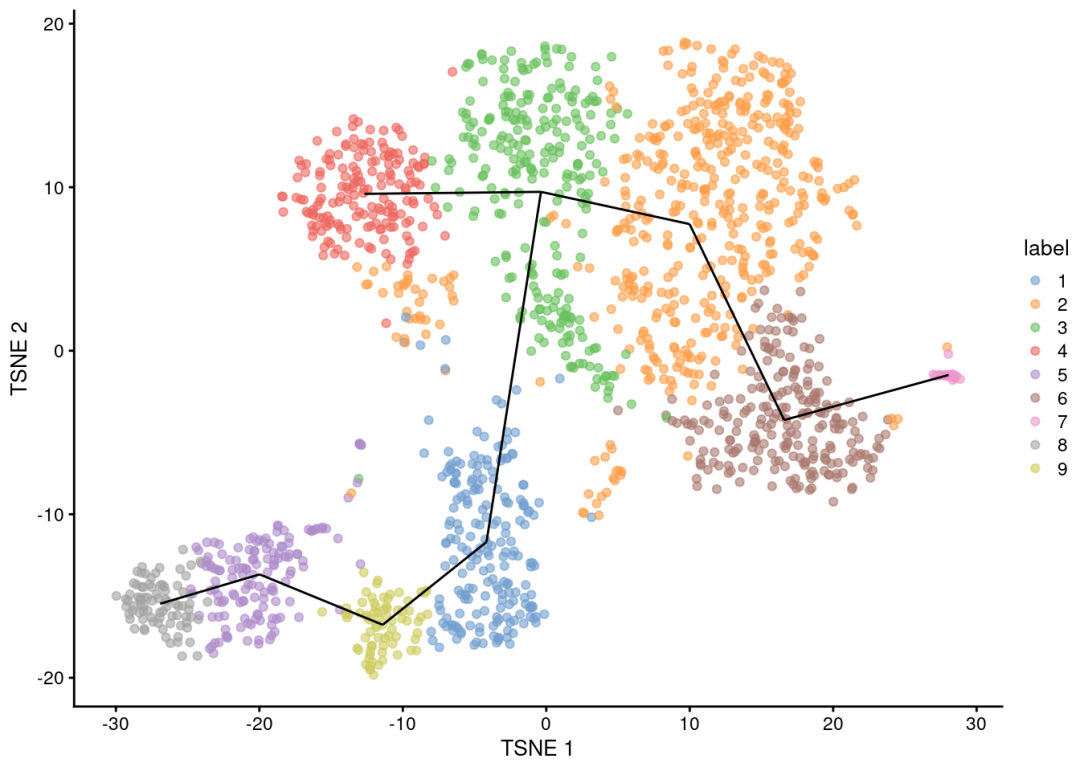
绘制显著差异表达基因的热图plotHeatmap(scRNA_data_corrected, genes = top_de_genes$gene_id)

绘制细胞类型间的基因表达差异箱线图plotGeneBoxplot(scRNA_data_corrected, genes = top_de_genes$gene_id, grouping_var = "CellType")
scuttle作为R语言生物信息学领域的一颗璀璨明珠,为单细胞RNA测序数据的分析和解释提供了强大的支持。借助scuttle,生物学家们可以更深入地挖掘细胞的多样性、发育过程和疾病机制,从而在生物学研究中迈出重要一步。
以上就是对R语言包的简单介绍啦,scuttle为生物学家和生物信息学家提供了一个强大的平台,用于处理、分析和解释单细胞RNA测序数据。通过这个包,研究人员能够深入挖掘细胞之间的复杂关系,发现隐藏在转录组数据背后的生物学信息。无论是在解码疾病的分子机制,还是在揭示生命的精彩多样性方面,scuttle都成为了不可或缺的工具,为生物学领域的进步贡献着巨大力量。
总的来说,scuttle作为R语言生物信息学领域的一颗明珠,为研究人员提供了一套强大的工具,用于探索单细胞RNA测序数据的世界。无论是揭示细胞的多样性、疾病机制,还是理解细胞的发育过程,scuttle都为生物学领域带来了巨大的推动力。通过其高效的分析和直观的可视化,scuttle不仅在解读数据中发挥着重要作用,也为我们展示了生命中细小而精彩的奥秘。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~
往期推荐
