从小白到空转达人:小果教你用Scanpy包玩转前沿热点空间转录组分析!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

大家好呀,我们又见面啦,今天和大家聊聊单细胞和空转分析中一个很基础的包——Scanpy,Scanpy 是一个Python包,用于分析与 AnnData(一种数据结构)联合构建的单细胞分析数据,当然目前随着技术的发展,它的开发者也在跟随时代热点变化不断改进它,现在除了单细胞分析,它也实现了空间转录组数据分析流程。之前小伙伴在后台问小果Scanpy和Seurat包的区别,说自己不是很懂各种分析流程的步骤和原理,果果今天就给大家详细讲讲python的scanpy包,其实scanpy和seurat的功能基本是可以替代的,不同的是一个用在python,一个用在R。
我们知道单细胞和空转分析流程包括很多步骤,比如质控(QC),标准化(Normalization),降维聚类(Dimensional reduction and clustering),Cluster marker genes, Spatially variable genes等,小果今天并不想直接给大家介绍网上都有的流程,而是想给大家看看Scanpy读取的数据结构,以及如何可视化,教大家一步入门Scanpy!
1.首先肯定是要输入文件!先来教教大家h5ad文件的读取
h5ad文件全称是h5anndata,是anndata的h5格式,anndata也是python中的数据格式,如果你想用seurat读,可以发现最后读取的文件格式会变为h5seurat,简单的说python中适配h5ad格式读取,R适配h5seurat格式读取。
adata =anndata.read_h5ad(‘F:/university/research/空间转录组/canvas/GSM6177599_10xVisium_processed.h5ad’)
2.读入文件后,让我们看一下adata中有什么
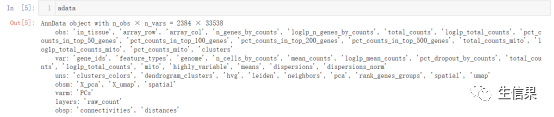
Obsm:对于观测的多维注释,它是可变的ndarray,就是说维度是可变的!小果偷偷告诉你,这里的维度一般是2至多维哦。而Obsm这里的m指的就是multi-dim多个维度的,obs_m对应于obs,但obs的每个成员都是一维的观测注释,obs_m的每个成员(X_pac和X_umap)都是多维的观测注释。哦对了,忘了告诉你,obs就是矩阵的行,也就是样本!
Varm:与obsm对应,是基因的多维观测~
uns:非结构化数据,也就是字典。
rank_genes_groups 识别差异表达的基因(潜在的marker),这个函数将获取每组细胞,并将每组中每个基因的分布与不在该组中的所有其他细胞中的分布进行比较最后进行识别。
pca:主成分分析降低数据维度。
umap降维:umap比tsne保留全局结构,允许大数据输入,运行时间短,这是umap和tsne的区别,一定要牢记喔!
obsp(pair):表示细胞和细胞之间的距离和连通性。
顺便给大家提提obs中的mito表示线粒体基因,一般mito都表示的是线粒体。
n_genes_by_counts`:每个细胞中,有表达的基因的个数;
total_counts:每个细胞的基因总计数(总表达量)
pct_counts_mt:每个细胞中,线粒体基因表达量占该细胞所有基因表达量的百分比
先给大家介绍这么多了,具体的还是需要后续自己查一下哦~
3.可视化一下
import matplotlib.pyplot as pltfrom matplotlib import rc_context# rc_context用于指定figure大小with rc_context({'figure.figsize': (4, 4)}):# vmax改变颜色最大值,frameon为去掉边框sc.pl.umap(Myadata, color=['Rp1','clusters','pct_counts_mito','total_counts'],s=50,ncols=4,frameon=False,vmax='p99')
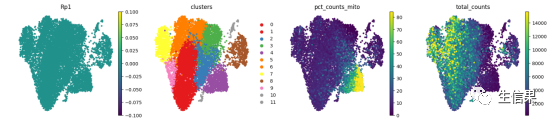
上图中我们使用umap绘制了对应的cluster,metadata的值,metadata就是每个细胞的信息喔,还绘制了Rp1,这么多图只需要简单的两行代码就可以绘制!是不是觉得写这个包的人很牛!
但是其实小果还是想知道这个坐标是哪里来的,所以就去试了一下adata中的obsm中的umap的值,想看看究竟在这个包中是不是X_umap的坐标绘制了上面这样的图,于是就进行了下述操作:
#测试一下umap 符合预期,确实是数据为细胞的坐标import matplotlib.pyplot as plt# 将二维数组转换为两个一维数组x和ydata=Myadata.obsm['X_umap']x = [d[0] for d in data]y = [d[1] for d in data]# 绘制散点图plt.scatter(x, y
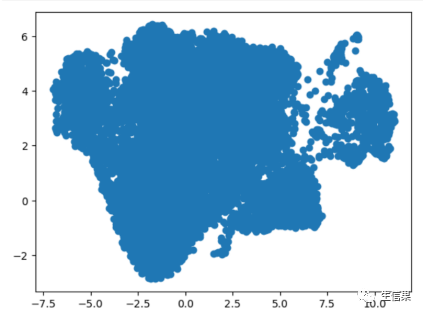
我们可以看到,确实是这样的!所以大家之后要是不懂包内部的原理的话一定要多尝试,多试试总归是骗不了人的!
with rc_context({'figure.figsize': (4.5, 3)}):'''stripplot=False用于删除internal dots, inner='box'用于在小提琴内部加方框图'''sc.pl.violin(Myadata, ['pct_counts_in_top_50_genes', 'total_counts_mito'], groupby='clusters', stripplot=False, inner='box')'''mino表示线粒体基因'''
可以再来试试小提琴图!
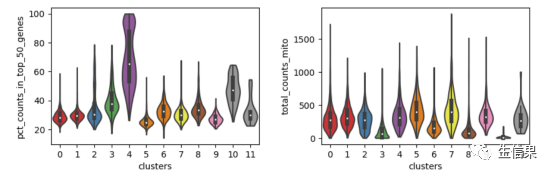
这是11个cluster中每个cluster对应的不同类的基因的小提琴图。
总结一下叭,pl.umap可以画umap降维图,pl.violin可以画小提琴图,那你知道热图怎么画吗,还有堆叠小提琴图,比如下面这种图所示,是不是很好看哈哈哈,小伙伴们可以之后试试哦,如果有需要的话,小果也会专门给大家出一期继续讲一下我们这次所用数据的后续分析的,就看大家的赞的个数了哦。
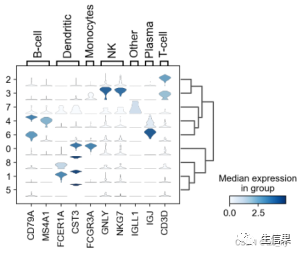
好啦~今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:(http://www.biocloudservice.com/home.html),其中也包括了通路表达分析(http://www.biocloudservice.com/313/313.php),单细胞的基因共表达分析(http://www.biocloudservice.com/906/906.php)等各种小工具哦~,有兴趣的小伙伴可以登录网站进行了解。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。
小果友情推荐
好用又免费的工具安利



点击“阅读原文”立刻拥有
↓↓↓