「付费」【代码-7】手把手带你学会SVM,轻松解决基因分类问题!
收录于话题
基因分类是一种广泛应用于生物信息学和医学领域的机器学习问题。在基因分类中,每个样本都由一个基因表达特征向量表示,而不同的样本对应于不同的病症类型,如癌症。
SVM算法可以通过学习一个分类超平面来将不同类型的癌症分开。在这里,超平面是一个n-1维的空间,其中n是特征向量的维数。当我们将超平面应用于基因分类时,每个基因表达特征向量都被看作是高维空间中的一个点。SVM算法的目标是找到一个最优超平面,使得不同类型的癌症分开,并且离超平面最近的样本点到超平面的距离最大化。这些最近的样本点被称为支持向量(support vectors)。通过最大化支持向量之间的距离,SVM可以更好地处理噪声和异常值,同时还可以避免过拟合。
简单来说使用SVM算法进行基因分类就像是在高维空间中寻找一个最优超平面,将不同类型的病症分类。这个过程就像是建立分叉路口,引导不同方向的行人走进不同的路口。而这个路口的位置和形状,就由SVM算法来决定。

在基因分类中,SVM算法通常使用径向基函数(RBF)核函数来将数据点映射到高维空间中。RBF核函数可以将低维空间中的数据点映射到高维空间中,使得我们可以使用超平面来分隔不同类型的癌症。通过调整RBF核函数的参数,我们可以控制分类器的复杂度和泛化性能。
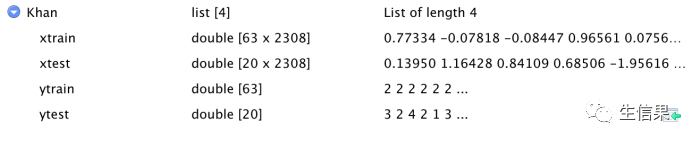
我们以ISLR包中的Khan数据为例。Khan是基因数据,每个观测对应于一个组织标本的数据, 这些标本分为4类,对应于4种小圆蓝色细胞瘤。变量xtrain和ytrain是训练样本,xtest和ytest是测试样本。
如果需要完整的代码可以点击付费获取哦!今天小果的分享就到这里,如果小伙伴有其他数据分析需求,可以尝试本公司新开发的生信分析小工具云平台,季代码完成分析,非常方便奥!
library(e1071)library(ggplot2)library(ISLR)#从ISLR包中加载名为Khan的数据集#输出Khan$ytrain的频率表,以了解数据集中的类别分布情况data("Khan", package = "ISLR")table(Khan$ytrain)#将训练集和测试集存储在train和test数据框中,其中xtrain和xtest是预测变量#ytrain和ytest是响应变量。在train和test数据框中,y变量被强制转换为因子变量train <- data.frame(x = Khan$xtrain, y=as.factor(Khan$ytrain))test <- data.frame(x = Khan$xtest, y=as.factor(Khan$ytest))
 自变量有2308个,所以自变量的空间维数很高,不需要用多项式或者径向基增加维度。可以直接用线性的SVM。
自变量有2308个,所以自变量的空间维数很高,不需要用多项式或者径向基增加维度。可以直接用线性的SVM。
(扫码领取整理好的输入文件,代码文件及示例结果)
