pySCENIC对单细胞转录因子分析可视化(下)
点击蓝字 关注我们
SCENIC(Single-Cell rEgulatory Network Inference and Clustering)是一个基于共表达和motif分析,用于计算单细胞转录组数据基因调控网络重建及细胞状态鉴定的方法,可以简单理解为单细胞转录因子分析。
官方教程https://github.com/aertslab/SCENIC
这里使用python版本的pyscenic和R可视化相结合的方法,利用单细胞注释完整的Seurat对象,得到不同组别和不同细胞类型中高表达的转录因子热图。
我们在上篇推送中得到了所需要的sample_SCENIC.loom文件,这里我们将其导入R,并结合Seurat对象的信息进行可视化。
首先加载需要的R包
library(tidyverse)library(Seurat)library(GENIE3) #基于共表达情况鉴定每个TF的潜在靶点,推断共表达网络library(RcisTarget) #基于DNA-motif 分析选择潜在的直接结合靶点,进行转录因子结合的motif分析library(AUCell) #鉴定具有激活基因集或基因网络的细胞,分析每个细胞的网络活动度library(SCENIC)library(SCopeLoomR) #读取loom文件需要用到#以上包安装的时候可能会遇到编译报错,大家后续如果有问题的话联系小果出教程哦library(RColorBrewer) #绘图library(patchwork) #绘图
导入数据
loom <- open_loom("sample_SCENIC.loom")接下来无脑运行就好,
有兴趣了解的可以去官网研究
regulons_incidMat <- get_regulons(loom, column.attr.name="Regulons")regulons <- regulonsToGeneLists(regulons_incidMat)class(regulons)regulonAUC <- get_regulons_AUC(loom, column.attr.name='RegulonsAUC')regulonAucThresholds <- get_regulon_thresholds(loom)tail(regulonAucThresholds [order(as.numeric(names(regulonAucThresholds)))])embeddings <- get_embeddings(loom)close_loom(loom)
导入Seurat对象
以上皮细胞为例,从Seurat对象(sce)中提取出来。要注意的是细胞数最好在一万以下,否则运行时长不可想象。epithe <- subset(sce, idents = "epithelial")sub_regulonAUC <- regulonAUC [,match(colnames(epithe),colnames(regulonAUC))]dim(sub_regulonAUC)identical(colnames(sub_regulonAUC), colnames(epithe))capture.output(regulons,file="TF2gene.txt")TFhigh <- names(regulons)write.table(TFhigh,file="TFhigh.txt")save(sub_regulonAUC,file="AUCellresult.RData")regulonActivity_byCellType <- sapply(split(colnames(epithe), epithe$celltype),function(cells), rowMeans(AUC[,cells]))regulonActivity_byCellType_Scaled <- t(scale(t(regulonActivity_byCellType), center = T, scale=T))))
保存一下
AUC <- getAUC(regulonAUC)colnames(AUC) <- epithe$groupAUC _sub <- AUC [rownames(AUC) %in% c("BATF(+)"……,] # TFhigh.txt里选出的TFdim(AUC_sub)
开始绘图
annotation_col = data.frame(CellType = epithe$celltype,Group = epithe$group)ann_colors=list(Group=c(N='blue',T='red')) #为不同组别标注不同的颜色pdf('SCENIC_heatmap.pdf', width =10, height = 3)pheatmap(AUC_sub,col= rev(brewer.pal(5, 'RdBu')),show_rownames=T,annotation_colors=ann_colors,annotation_col = annotation_col,cutree_col=3)dev.off()
这样就完成高表达的转录因子热图的可视化啦~让我们看看
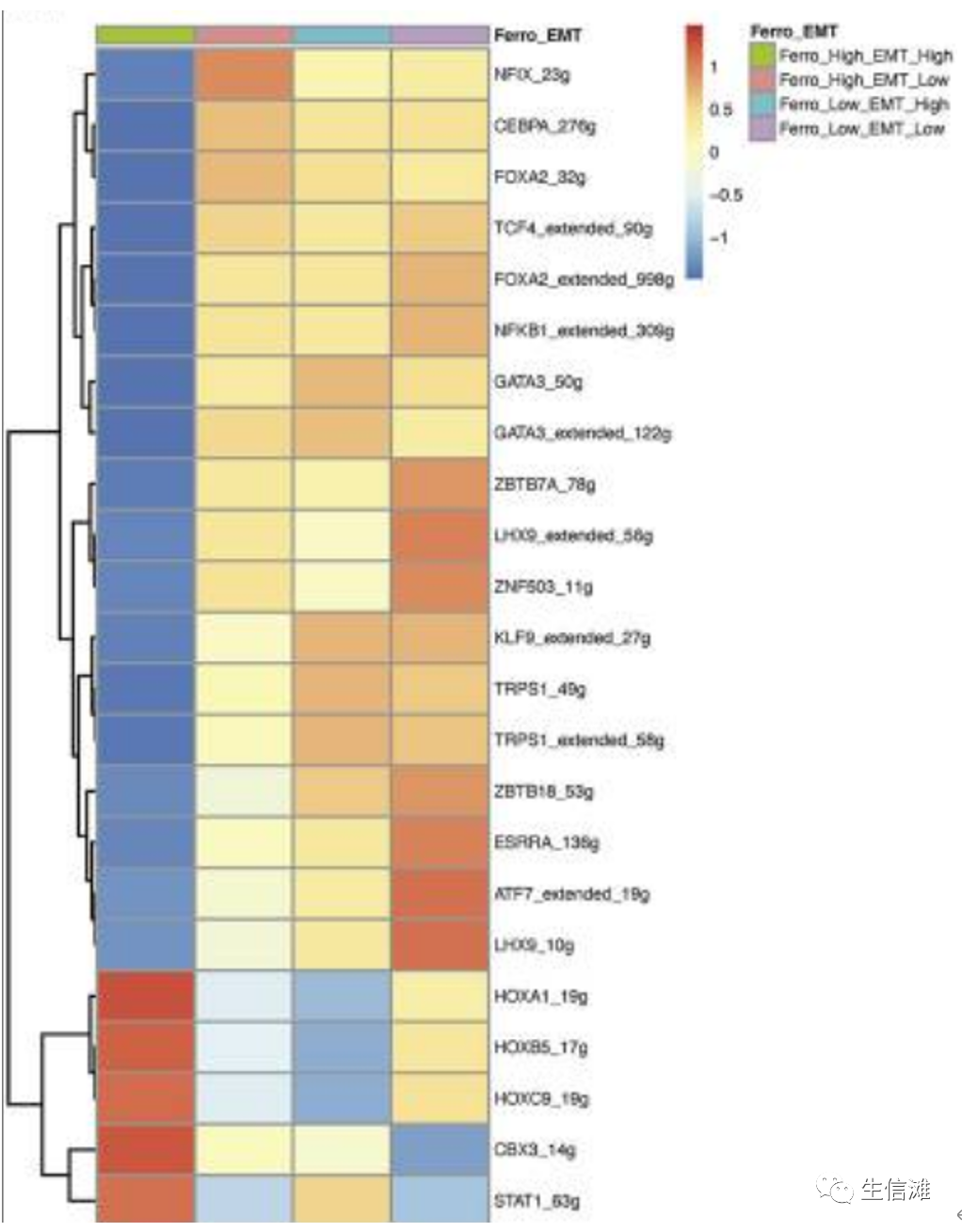
#图片来源:Yao J, et al. Single-Cell RNA-Seq Reveals the Promoting Role of Ferroptosis Tendency During Lung Adenocarcinoma EMT Progression. Front Cell Dev Biol. 2021
前面我们提到,SCENIC占用大、耗时长,而小果这次介绍的pySCENIC速度比R版本的有十分显著的优势!学会pySCENIC让你快人一步!
/2_h5YsicpkVJhg8kTg4kufoaoicoUpEQ.png)
点击“阅读原文”进入网址