保姆级教程!小果教你如何使用DAVID数据库并可视化结果!
生信人R语言学习必备
立刻拥有一个Rstudio账号
开启升级模式吧
(56线程,256G内存,个人存储1T)

小伙伴们在做GO功能富集和KEGG通路富集分析时候,遇见过很多困难,其中之一就是在用R做KEGG分析时候,KEGG数据库因为网络进不去导致KEGG通路富集失败。还有的小伙伴想做富集分析,但是只有基因名,少了logFC值,导致想做富集分析很苦恼。小果在这里教大家一个方式,使用DAVID数据去分析。
目前来说DAVID数据库在2022年更新了,那就意味着我们又可以使用了,之前因为长时间没跟新,导致许多的数据跟不上时代。现在小果带大家手把手教大家如何使用DAVID数据去做富集分析!
我们先进入数据库官网(https://david.ncifcrf.gov/)
上面有很多的功能,我们只需要点击start Analysis就可以了
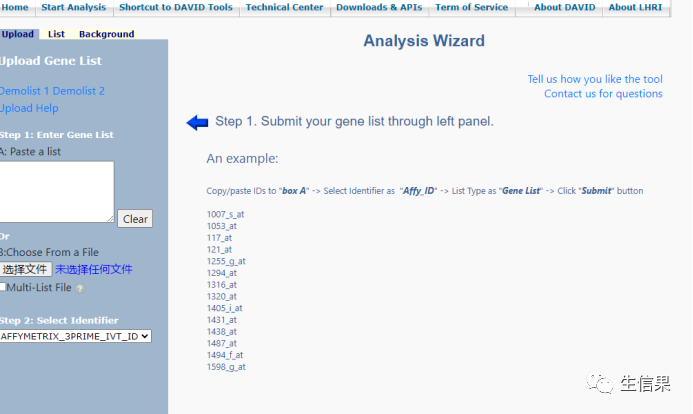
在框内输入我们想要分析的基因名或者导入文件,小果在这里给大家演示,使用的示例数据。
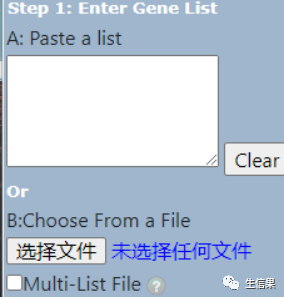
接下来要注意我们在第二步要选择“OFFICIAL_GENE_SYMBOL”因为我们的基因是通过SYMBOL转换过来的,小伙伴不要选错了

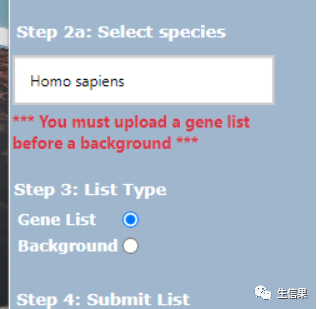
接下来选择我们的人类,以及第三步【类型】是基因列表。然后我们开始分析吧 ,点击Submit List
等待几秒钟:
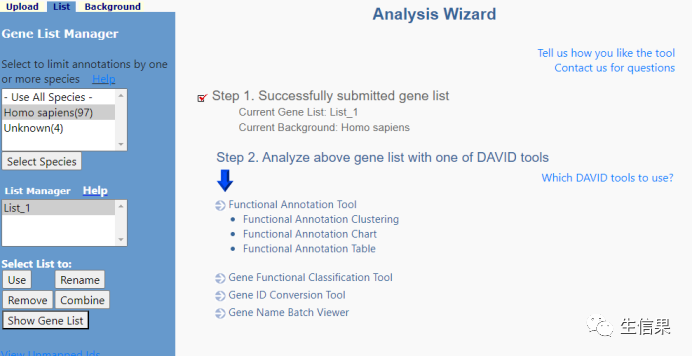
就出现这样的界面,右侧是在人类数据库中找到了97个Gene我们只要这97个,Use它
然后我们查看富集分析结果:
点击这个:

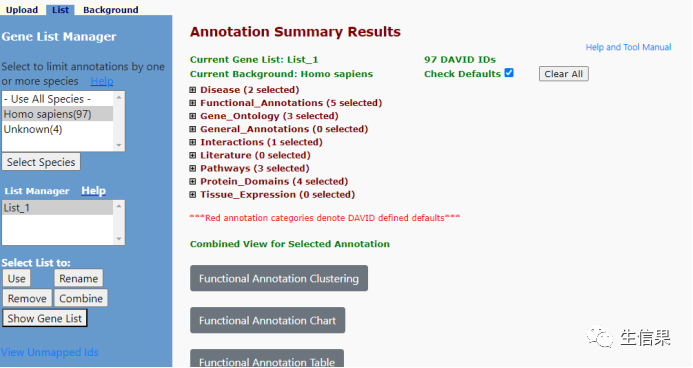
我们查看结果我们先打开通路富集结果:

KEGG通路是我们需要的,我们点击KEGG结果的Chart:
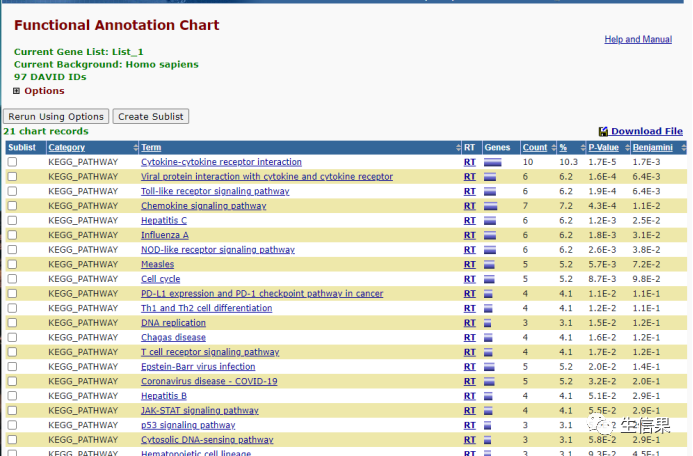
然后我们点右上角的下载
但是下载的结果是
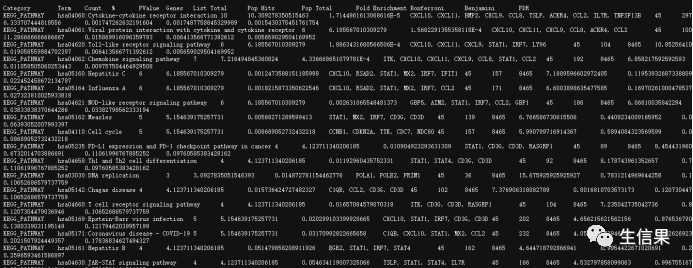
这就需要我们新建一个txt文件,我们把结果保存进去
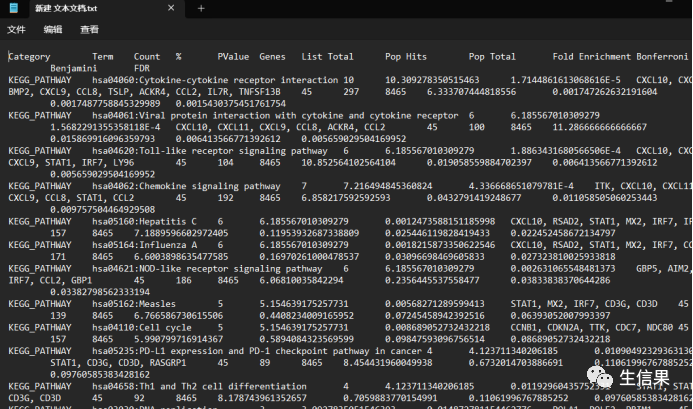
小伙伴自行保存,可以看到富集的通路以及其他信息。
接下来我们去可视化KEGG富集的结果。
我们先读入数据:
library(stringi)library(ggplot2)library(dplyr)##读取和整理KEGG的结果downgokegg<-read.delim("E:\生信果\KEGG.txt")接下来对数据处理,选择需要可视化的数据:enrich<-downgokeggenrich_signif=enrich[which(enrich$PValue<0.05),]enrich_signif=enrich_signif[,c(1:3,5)]head(enrich_signif)enrich_signif=data.frame(enrich_signif)KEGG=enrich_signifKEGG$Term<-stri_sub(KEGG$Term,10,100)
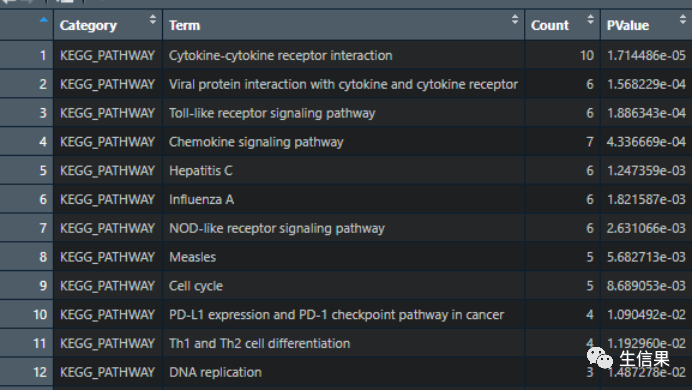
最后可视化:ggplot(KEGG,aes(x=Count,y=Term))+geom_point(aes(color=PValue,size=Count))+scale_color_gradient(low='slateblue4',high='firebrick3')+theme_bw()+theme(panel.grid.minor = element_blank(),panel.grid.major = element_blank())
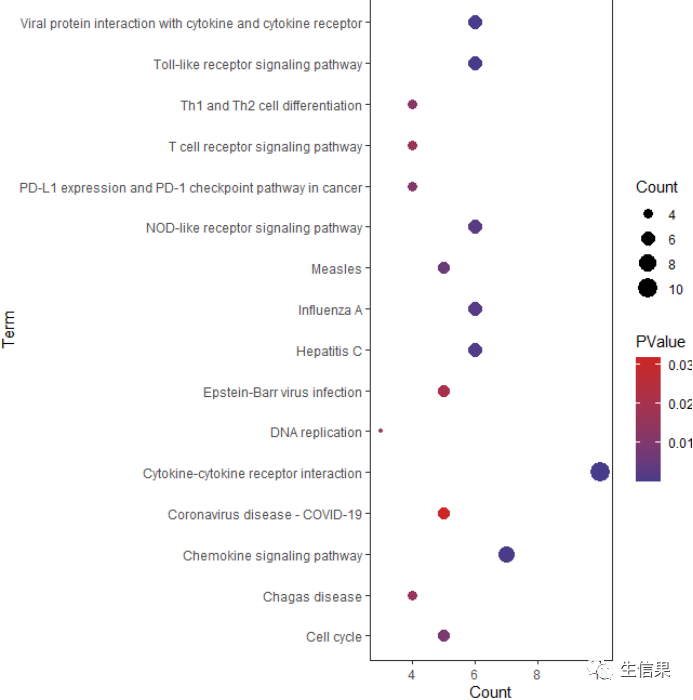
我们以气泡图的形式展示,以Count值为横坐标。
接下来我们去分析GO富集结果
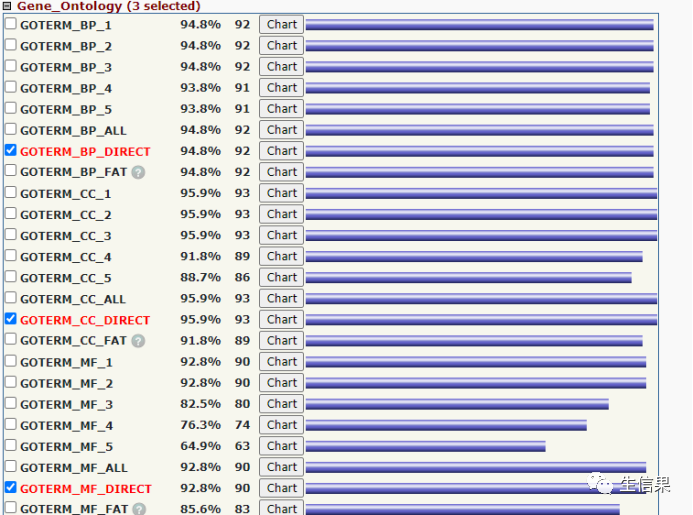
我们在这里找到GO富集的结果,其中有BP,CC,MF。和上述KEGG结果一样把结果保存到txt文件中。小果在这里提前保存好了,小伙伴自行去保存哦。

把结果分成三个文件夹。接下来我们去分析吧:我们把数据一一导入进去,然后在合并数据
GO_CC<-read.delim('E:\生信果\CC.txt')GO_CC_signif=GO_CC[which(GO_CC$PValue<0.05),]GO_CC_signif=GO_CC[,c(1:3,5)]head(GO_CC_signif)GO_CC_signif=data.frame(GO_CC_signif)GO_CC_signif$Term<-stri_sub(GO_CC_signif$Term,12,100)GO_BP<-read.delim('E:\生信果\BP.txt')GO_BP_signif=GO_BP[which(GO_BP$PValue<0.05),]GO_BP_signif=GO_BP_signif[,c(1:3,5)]head(GO_BP_signif)GO_BP_signif=data.frame(GO_BP_signif)GO_BP_signif$Term<-stri_sub(GO_BP_signif$Term,12,100)GO_MF<-read.delim('E:\生信果\MF.txt')GO_MF_signif=GO_MF[which(GO_MF$PValue<0.05),]GO_MF_signif=GO_MF_signif[,c(1:3,5)]head(GO_MF_signif)GO_MF_signif=data.frame(GO_MF_signif)GO_MF_signif$Term<-stri_sub(GO_MF_signif$Term,12,100)enrich_signif=rbind(GO_BP_signif,rbind(GO_CC_signif,GO_MF_signif))go=enrich_signifgo=arrange(go,go$Category,go$PValue)对于读取的数据小伙伴可以在参数设置选择自己想可视化的数据结果。接下来我们设置一下图例的名称:##图例名称设置m=go$Categorym=gsub("TERM","",m)m=gsub("_DIRECT","",m)go$Category=mGO_term_order=factor(as.integer(rownames(go)),labels = go$Term)COLS<-c("#66C3A5","#8DA1CB","#FD8D62")
有的小伙伴想用Count值去展示,也是可以的,看小伙伴的需求。最后我们开始画图:
###开始画图ggplot(data=go,aes(x=GO_term_order,y=Count,fill=Category))+geom_bar(stat = "identity",width = 0.8)+scale_fill_manual(values = COLS)+theme_bw()+xlab("Terms")+ylab("Gene_counts")+labs()+theme(axis.text.x = element_text(face = "bold",color = "black",angle = 90,vjust = 1,hjust = 1))
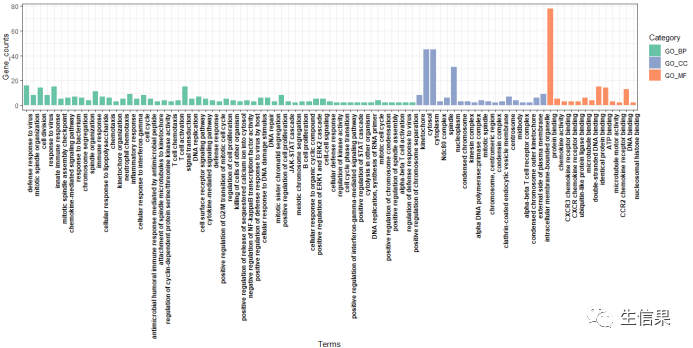

这样我们的结果就展示出来了,包含BP CC MF 我们以柱状图的形式展示,不过有的小伙伴想以其他的形式,比如气泡之类,小果在这里就不一一教学了。感兴趣的小朋友可以自行去设置。最主要的我们的结果已经出来,小伙伴可以根据结果去可视化。
快去试试吧!
其他相关分析内容,例如预测肿瘤样本药物敏感性分析(http://www.biocloudservice.com/712/712.php),预测某样本亚型对免疫治疗的反应(http://www.biocloudservice.com/292/292.php),单样本富集算法分析免疫浸润丰度(http://www.biocloudservice.com/106/106.php),计算64种免疫细胞相对含量(http://www.biocloudservice.com/107/107.php)等都可以用本公司新开发的零代码云平台生信分析小工具,一键完成该分析奥,感兴趣的小伙伴欢迎来尝试奥,网址:http://www.biocloudservice.com/home.html。今天小果的分享就到这里,下期在见奥。
小果友情推荐
好用又免费的工具安利



点击“阅读原文”立刻拥有
↓↓↓