看这里,三分钟教会你使用R语言msa包,让序列比对不再是难题!!

公众号后台回复“111”
领取本篇代码、基因集或示例数据等文件
文件编号:240314
需要租赁服务器的小伙伴可以扫码添加小果,此外小果还提供生信分析,思路设计,文献复现等,有需要的小伙伴欢迎来撩~

if (!requireNamespace("Bioimager", quietly=TRUE)) install.packages("BiocManager")BiocManager::install("msa") # 安装测试msa包是否安装成功,输入library(msa) # 加载R包packageVersion("msa") # 查看R包的版本

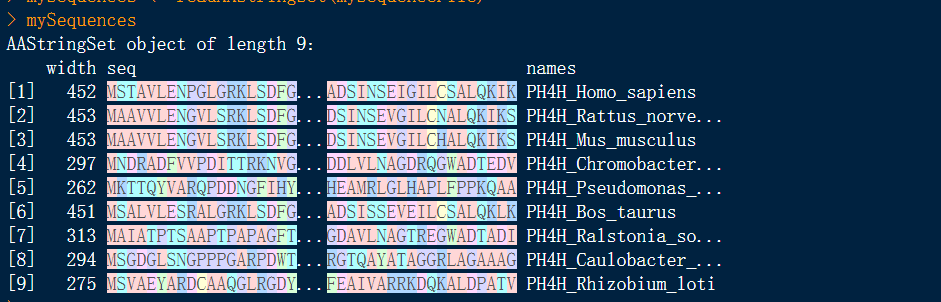
mySequenceFile <- system.file("examples", "exampleAA.fasta", package="msa") #加载文件mySequences <- readAAStringSet(mySequenceFile) # 读取到变量中mySequences #显示结果
myFirstAlignment <- msa(mySequences) #多序列比对
myFirstAlignment # 显示多序列比对结果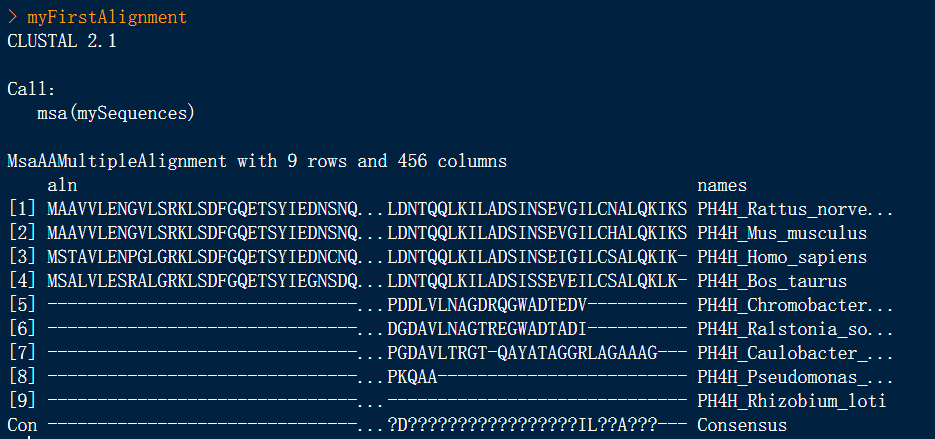
print(myFirstAlignment, show="complete") # 显示完整结果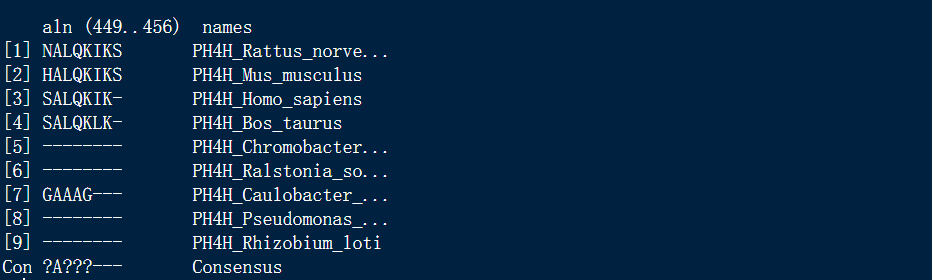
msaPrettyPrint(myFirstAlignment, output="pdf", showNames="none", showLogo="none", askForOverwrite=FALSE, verbose=FALSE) 
Class(myAlignment) # 查看数据格式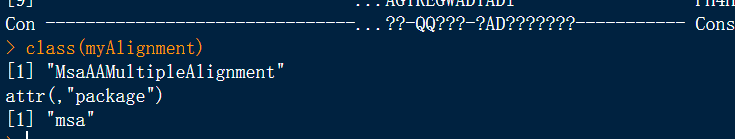
library(seqinr) # 加载seqinr包myAlignment2 <- msaConvert(myAlignment,type="seqinr::alignment") # 数据格式转换class(myAlignment2) # 查看数据格式

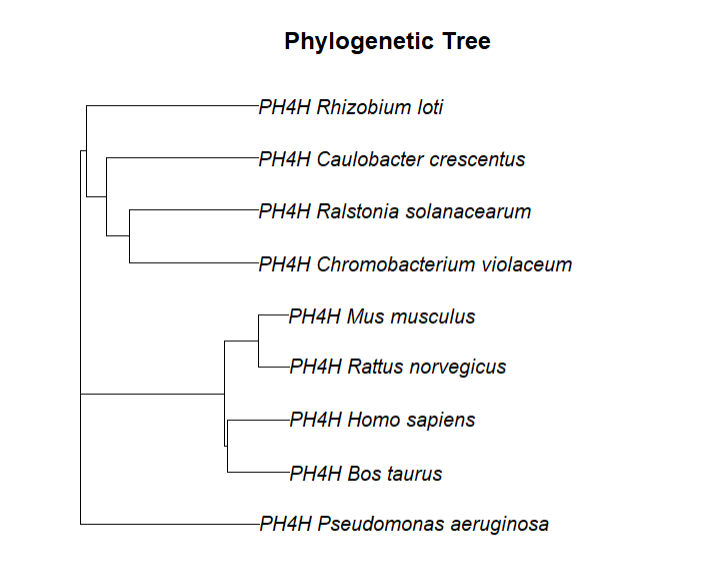
d <- dist.alignment(myAlignment2, "identity") # 计算比对距离library(ape) # 加载ape包hemoTree <- nj(d) # 根据计算的比对距离构建系统发育树plot(hemoTree, main="Phylogenetic Tree") # 将构建的系统发育树展示出来
小果还提供思路设计、定制生信分析、文献思路复现;有需要的小伙伴欢迎直接扫码咨询小果,竭诚为您的科研助力!

定制生信分析
服务器租赁
扫码咨询小果


往期回顾
|
01 |
|
02 |
|
03 |
|
04 |